Keywords
stress / inducible response / Pseudomonas fluorescens / bacteriophages
stress / inducible response / Pseudomonas fluorescens / bacteriophages
We made numerous edits to the manuscript to address the points raised by the two referees. Notably, we gave more details about our experimental protocol, and the different steps taken to ensure that our results are not artifacts. We discuss in more depth the potential mechanisms that can trigger the effect we describe, and suggest further studies to clarify its adaptive role.
See the authors' detailed response to the review by Britt Koskella
Pathogens are ubiquitous in natural communities1 and the antagonistic interactions they establish with their hosts are recognized as one of the main drivers of evolutionary diversification2,3. Hosts can reduce the impact of pathogens through three non-mutually exclusive processes4: (i) avoidance of either infected individuals, habitats where the pathogen is prevalent, or of the pathogen itself5, (ii) resistance to the actual infection process or post-infection immune defences6, and (iii) tolerance7. Research on these responses has generally focused on animal and plant models, but there is growing appreciation that microbes, particularly bacteria, can exhibit similar responses. For instance, bacteria can be selected for heightened levels of genetic resistance towards infection by pathogens8–10. On the other hand, although bacteria are known to display plastic responses to various types of environmental stresses11,12 and to competition13, it is unknown whether they can do so when faced with natural enemies such as bacteriophages.
Plastic responses are an adaptive phenotypic change following an environmental stimulus, occurring without a concurrent change in the genotype14. They may involve behavioural, physiological or phenological changes15,16, and be triggered by direct or indirect contact with the stimulus17 or through communication with neighbouring organisms18. Phenotypic plasticity is considered to be a genetic adaptation to variable environments, but given the diversity of associated mechanisms and behaviours, it is not known to what extent different stimuli translate into different responses15,19.
Individual-level interactions between bacteria and phage may be conducive to induced responses. The first step of bacteriophage infection is the binding of phage proteins to bacterial surface proteins20, which then triggers conformational changes to both proteins21. Surface proteins used by the bacterium for signal transduction are known to be targets of bacteriophage adsorption22 and as such could trigger a response when bacteriophage binding is detected. Such a response would allow a bacterium to react to the pathogen and to eventually either evade or reduce the effects of the infection. Lytic phages are prime candidates for organisms against which bacteria may have evolved a stress response, because they typically interact with their host over short timescales, and death is inevitable once the phage has injected its DNA into a sensitive bacterial cell.
In addition, bacteriophages are widely distributed in the environment20 and interact with their hosts over relatively small spatial scales23 and throughout most of the year24,25. This could select for the expression of induced structural, physiological or behavioural responses to different enemies. Also, bacteria employ signalling pathways and have a known ability to communicate within populations26. Such pathways could induce and synchronise inducible responses before predators and pathogens are encountered, or at least before they have spread through the population, or before the point beyond which cell death is certain. All of these factors suggest that plastic stress responses to phage should be a common feature of bacterial cells and that such responses would have important repercussions for ecological and evolutionary interactions between phage and bacterial populations. Although molecular responses of bacteria to bacteriophages have been characterized27, the behavioral, ecological, and selective consequences of such responses are not known.
Here we demonstrate that when confronted with phage, bacteria express transient increases in division rate at a cost to individual biomass accumulation28. Specifically, we employ the rhizosphere bacterium Pseudomonas fluorescens SBW2529 to investigate how its population growth rate is affected by exposure to inactivated populations of is lytic bacteriophage SBW25Φ230. We find that bacteria exposed to inactivated phage increase their fission rate nearly two-fold at 24 hours post-exposure. This is followed by a continual decrease in fission rate relative to the control. We also show that bacteria exposed to inactivated phage were smaller in size compared to controls. By the end of the experiment, bacteria regained their original growth rate, but not their original size, which implies differences in energy allocation constraints between these two life-history traits. All of these effects were enhanced as the density of inactivated phage was increased. The results are consistent with a behavioural strategy that increases allocation to reproduction under stressful conditions (i.e., “terminal investment”). Terminal investment is well characterised for other host-parasite associations31, but to our knowledge has not previously been observed in bacteria subject to phage infection.
Bacteria exposed to UV-inactivated phage display a statistically significant higher growth rate over the first 24 hours post-exposure than non-phage controls (Kruskal-Wallis, df = 3, P = 0.006; Figure 1). After this period, the estimated doubling time of exposed bacteria increased (i.e., their populations grew slower), and did so for the next 48 hours. This decrease in growth rate compared to controls is suggestive of a cost to the higher fission rate observed over the first 24 hours (Figure 1). During the fourth day post-exposure, control and treatment bacteria showed no significant differences in doubling time (KW, df = 3, P > 0.05). That exposed bacteria returned to their ancestral growth rate suggests that the response over the first 24 hours was due to phenotypic plasticity and not selection on faster growing genotypes. There was a marginally significant effect on population growth for bacteria exposed to different phage concentrations (KW, df = 2, P < 0.02), suggesting that the encounter rate between bacteria and phage is important in determining the population-level strength of the fission response.
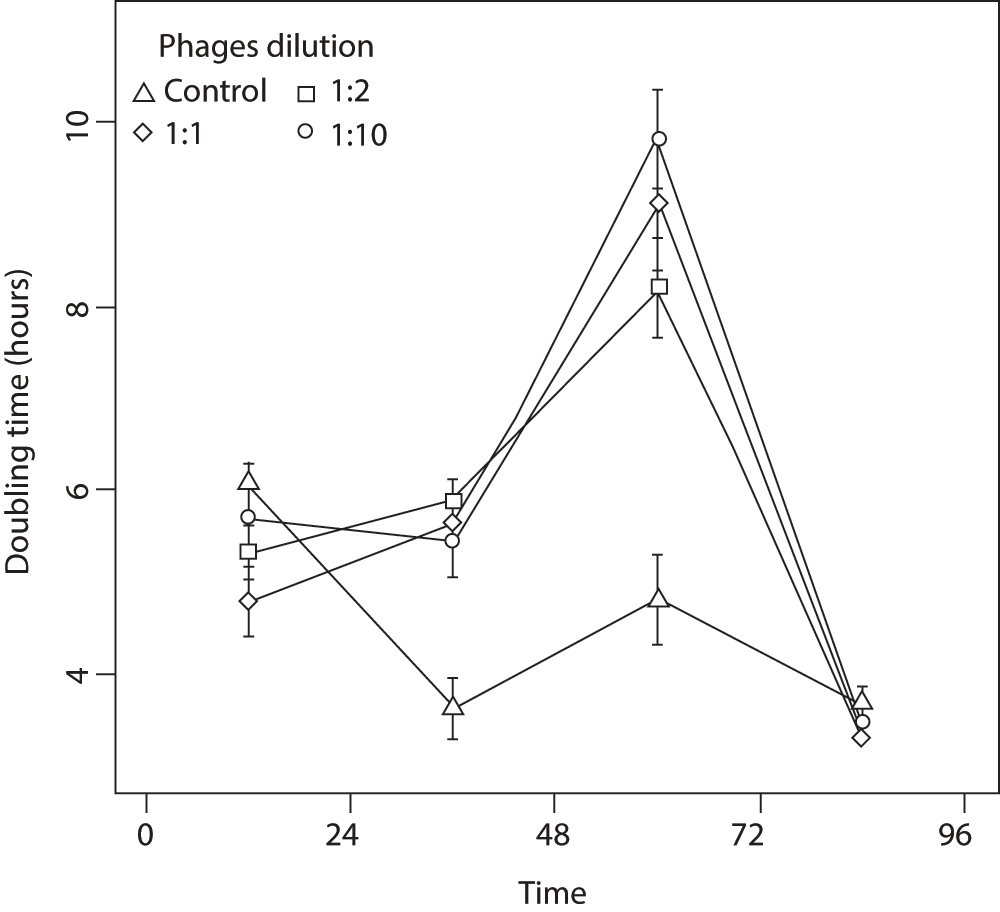
This was measured for four consecutive days following four hours exposure. Bacteria exposed to phage grew significantly faster than controls over the first day, and then expressed an apparent cost in terms of smaller cell size that attenuated by the fourth day. Central points are the means of 12 replicates, and the bars are standard errors.
We hypothesized that faster doubling times would come at a cost to cell size, since cells would have less time to metabolize and convert absorbed nutrients into cell structure twenty-four hours post-exposure, we found that phage-treated bacteria were two to three times smaller (as measured by mean cellular width) than the control (KW, df = 3, P < 0.0001; Figure 2). This difference in size gradually decreased over the following 3 days, but in contrast to growth rate (Figure 1), bacteria did not attain their ancestral cell size by the end of the experiment (Figure 2). Analyses of the distribution of several flow cytometry profiles showed that a difference in cell shape is unlikely to explain this result (see Data File below). Namely, whereas the bacterial populations differed with regards the side scatter parameter, forward scatter showed no change in its distribution. This implies that bacterial shape remained unchanged throughout the experiment, and indeed, additional observations using a transmission electron microscope showed that the cells remained rod-shaped for all treatments.

We did not observe any difference in the impact of live phage on bacterial populations exposed to the different treatments (KW, df = 2, P = 0.153), suggesting that the inducible response does not alter bacteria resistance to phage predation.
Our experiments reveal a previously unexplored behavioural response to bacteriophage predation: phage induce bacteria to reproduce earlier in their cell cycle. We hypothesize that this response increases the survival chances of bacterial progeny under natural conditions and demonstrate that this behaviour comes at a fitness cost of reduced size of daughter cells. Our experiments with UV-inactivated phage further demonstrate that this response is specifically due to phage binding. An alternative explanation is that phage binding decreases resource uptake by bacterial cells. However, this seems unlikely in our experiment. Because bacteria were exposed to inactivated phages only, the total number of viral particles is predicted to stay constant (or possibly degrade) throughout the experiment. When bacteria divide, the number of phages bound to a daughter cell should be roughly half the number on the mother cell; thus, the number of bound phages per cell will decrease exponentially with cell divisions. Using the density of phages and bacteria employed in our experiment, we predict that there will be, on average, less than one phage individual per bacterial cell after 9 to 10 cell divisions, which based on the mean doubling time presented in Figure 1, is reached in the first 48 hours of the experiment. Our results can explain previous observations on phage-associated increases in population size in P. fluorescens32. Specifically, we predict that a significant number of phage in the experiments of Gomez and Buckling32 did not kill their bacterial hosts before some of the latter were able to accelerate their cell cycle and produce daughter cells. Furthermore, our results support and extend both theoretical33 and empirical34,35 predictions that victims may lessen the fitness impact of their natural enemies through early reproduction, to cases where phenotypic responses are plastic and temporary. Increased allocation to reproduction in stressful environments–termed “fecundity compensation” or “terminal investment”31–although never studied in bacteria-phage associations to our knowledge–has been extensively studied for other host-parasite (or organism-stressor) interactions. Terminal investment is characterized by increased reproductive rate or the earlier onset of reproduction, if the prospect of future reproduction is low36. Examples of such responses include faster host maturation37, increased oviposition rate38, and the modification of traits involved in the onset of reproduction39,40. This response is expected to result in smaller individual size, because energy allocated to growth is directed to reproduction when the stressor is present.
Phenotypically plastic responses are important in that they allow individuals to cope with environmental change during their lifetimes41. As such, plasticity is expected to be favoured in variable environments when the costs of induction and phenotypic change compensate for probabilistic (expected) fitness loss42. Although it is difficult to generalize about constitutive costs of resistance across biological systems43,44, limited evidence suggests that genetically evolved, constitutive resistance in bacteria to their lytic phage could have costs of as much as 5–10% to relative fitness45.
We employed inactivated bacteriophages to evaluate how phage contact with the bacterial outer membrane mediates bacterial responses. Bacteria could be selected to exhibit an escape response in several, non-mutually exclusive ways. First, non-virulent phage may signal the presence of virulent phage in the local environment (i.e., the bacterium does not perish following initial phage contact). Senescent (inactive) phage are present in natural environments25, and many phages bind to outer membrane proteins without being infective (e.g. the bacterium is resistant;45). Moreover, it is possible that phage could detach if they sense the host to be unsuitable46. Second, when phage infect the bacterium there may be a ‘race’ between the time it takes a bacterial cell to divide (and potentially survive) and the point of no recovery associated with the maturation of phage progeny and bacterial cell lysis. Third, the response may be a consequence of lysogens competing with lytic phages for host exploitation; the latter could benefit from early host reproduction in the presence of lytic competitors. However, sequencing of the P. fluorescens SBW25 genome revealed a low abundance of prophage-like regions47.
We were not able to determine whether the bacteria or the phage benefit from faster bacterial reproduction, and the literature reports effects both of facilitation and decrease in host metabolism upon infection48. Previous theoretical work suggests that phage productivity increases in bacteria with short life-cycles49. This is supported by recent empirical study employing the same strain of P. fluorescens50. Assuming that the physiological mechanisms involved in fission rate increases are the same in the two experiments, this suggests that rapid multiplication is not adaptive for the bacterium, and indeed we report no advantage of being exposed to inactived phage in terms of a lessened population impact during live phage exposure. Upon exposure to phage, bacteria reproduce faster, but experience a persistent reduction in individual size. Smaller cells have less surface area, and assuming that the density of receptor proteins does not change with cell size, this suggests that they will have lower encounter rates with phage. One possibility is that cell division allows bacterial cells to concentrate phage in one of the daughter cells51,52, resulting in some progeny managing to escape the pathogen. Future studies should therefore focus on the possible adaptive nature of this response for both bacterium and phage, by investigating in greater depth how it affects the mechanisms of infection, recovery, and resistance.
Ancestral Pseudomonas fluorescens SBW2529 were inoculated into 30 ml microcosms containing 6 mL of King’s B medium (KB), and allowed to grow under alternating rotational agitation (200 rpm for 1 minute every 30 minutes). Every 48 h following plating on solid agar, 10 CFU of the smooth morphotype were transferred into fresh KB medium. After 10 transfers, the culture was composed of smooth morphotypes only. We continued this selection procedure for another 10 transfers and then arbitrarily isolated a single CFU, which was used for all experiments described below. Experiments were conducted at 28°C in KB medium under constant rotational agitation (200 rpm).
We grew an arbitrarily selected clone of the ancestral phage SBW25Φ230 on an exponentially growing culture of fixed smooth P. fluorescens SBW25 in 3 mL of KB for 48 hours. This resulted in a culture containing approximately 108 phage per ml. The sample was then centrifuged for 3 minutes at 8000 rpm in a 1.5 ml Eppendorf tube, and the pellet discarded. Centrifugation was repeated three times to ensure all bacteria were removed (see Supplementary Figure S1). Phages were then isolated by centrifuging the remaining supernatant for 8 minutes at 13000 rpm, and inoculating the pellet into fresh KB medium. The sample was thoroughly vortexed and exposed to UV light (Model 4.LC, Vilber Lourmat, Deutschland, 254 nm wavelength) at 5 cm distance for 4 hours. Extensive pilot studies demonstrated that this method was sufficient to kill all phage (see Supplementary Figure S2).
We conducted a series of preliminary tests to verify how UV-inactivated phage affected bacterial hosts. First, observations under a transmission electron microscope showed that UV-inactivated phage were still intact and able to bind to their bacterial hosts. Second, we checked that bound UV-inactivated phage did not introduce phage DNA into the bacteria. This was done by inoculating 1 ml of UV-inactivated phage into 6 overnight bacterial cultures. Inactivated phage were allowed 4 h to attach to the bacterial outer membrane. We separated phage and bacterial fractions by filtration using a 0.2 µm filter. We then conducted a full DNA extraction (WholeBlood NucleoSpin DNA extraction kit, Macherey-Nagel) of the filter. PCR was done using TPV1f (GATGTGAGAAAGCGATACACGG) and TPV1r (GAGAGAAGCGGGAGAGTGAA) sequences developed for this study, which selectively amplify a 550 bp fragment of the phage DNA and a 1200 bp fragment of the bacterial DNA (see Supplementary Figure 1 for detailed protocols). We did not find any evidence that UV-inactivated phage was present in samples putatively containing bacteria only, thus confirming that (i) the DNA of inactivated phage was not incorporated in the bacterial cell and (ii) our centrifugation method removed both bound and unbound phage. Observations of c. 50 cells using TEM (Zeis EM10) showed no bound phages after the centrifugation treatment.
We conducted an experiment to understand how UV-inactivated phage affected bacterial behaviour. Fixed SBW25 bacteria of the smooth morphotype were first cultivated in 6 ml KB in 30 mL universal glass vials. 20 µL of exponentially growing bacteria (c 104 bacterial cells) were transferred into fresh KB medium with either no phage or UV-inactivated phage at ratios of 1:10, 1:2, and 1:1 (corresponding to approximately 106, 5×106, and 107 phage per ml), and then allowed to interact for 4 hours under alternating shaking (200 rpm for 1 minute every 30 minutes). KB medium containing UV-inactivated phages was obtained through centrifugation of inactivated phage, which were further added into pure KB, so that the medium used in the treatments only differs from the control by the presence of phages. Bacteria were then separated from bound phage by centrifuging (see above) and placed in fresh KB medium. 1% of each population was transferred every 24 hours into new KB medium. Each of the 4 treatments was replicated 6 times and arranged arbitrarily in a rack for incubation.
Biomass doubling time (used as a proxy for population fitness) was measured in a Fluostar Optima spectrophotometer (28°C, constant agitation, 250 measures at 650 nm over 24 hours) each day, using the following formula:
(1) Dt = [∆t ln(2)]/[ln(N*) – ln(N0)]
where N* and N0 are the total biomasses (measured as optical density, OD) before and after the exponential growth phase, and ∆t is the duration of the exponential phase. Exponential phase was determined by conducting a series of windowed linear regressions over the full growth curve, and retaining the part of the curve with the largest slope (computer code given in Supplementary materials part 3).
Individual cell size was measured by flow-cytometry using a FacsCantoII (BD BioSciences, San Jose, California, USA), and data (forward scatter) were analysed using the flowCore package53 in R 2.12.054. Each measure was performed on a sample of 2×105 cells without dyes.
Measures of OD will be affected by changes in particle size. At equal bacterial density, a population of smaller cells will yield a lower OD value, because fewer particles will block less of the incoming light. The practical conclusion is that whenever bacteria get smaller, we understimate their count, and thus their growth rate. Because this means that we are more conservative about the impact of phage exposure on growth rate (i.e., if there were any bias in our results, it would be an underestimation of the increase in growth rate), we did not correct for this effect.
We also estimated the sensitivity of the different treatments to live phage by measuring changes in bacterial populations. At each 24-hour transfer, 1% of the bacterial population was placed in 2 mL of fresh KB, and 20 µL of amplified phage (ca 108 viral particles) were added (a control without phage was conducted simultaneously). Bacteria CFUs were counted on solid agar after 48 hours of incubation to estimate population size.
Due to non-normality of the data as assessed by a Shapiro test, we used a Kruskal-Wallis test to determine the significance of the between-treatments effects.
TP, TB and MEH designed the research, TP and EM conducted the microbiology experiments, TP and CGB conducted the molecular biology experiments, TP, TB and MEH analyzed the results and wrote the paper, all authors contributed to revisions.
TP is funded by a CNRS-Région Languedoc Roussillon doctoral grant. TB was funded by NERC. MEH is funded by ANR EvolStress (ANR-09-BLAN-099-01), by the Ec2Co “Cytrix” program, and the McDonnell Foundation (JSMF 220020294/SCS-Research Award).
We are indebted to Cédric Mongellaz for his assistance with flow cytometry, and the Montpellier RIO Imaging center for providing technical support. We thank Michael Brockhurst and Angus Buckling for discussions.
PCR cycle – 6 minutes at 95°C, then 30 cycles of 1 minute at 94°C, 1 minute at 55°C, 2 minutes at 72°C, then 10 minutes at 72°C.
PCR buffer – 5 μL of buffer, 4 μL of primers at 10 pM/mL (for both TPV1f and TPV1r), 2 μL of dNTP at 5 pM/mL, 1.5 μL of MgCl2 at 25 mM, 5.35 μL of H20, 3 μL of sample DNA, 0.15 μL of TaqPol - conducted with a GoTaq FlexiDNA Polymerase M8301 kit from Promega.

The primers TPV1f and TPV1r yield a 1200 bp amplicon in the bacteria, and a 500 bp amplicon in phages. Our seperation method for bacteria and phage was complete, since only DNA of the intended organism was found in any given sample.
We verified the efficiency of the phage inactivation protocol by incubating the bacterial strain used for the main experiment with either live phage or phage exposed to UV for 2hrs or 4hrs. We measured the Malthusian fitness of 6 host populations near carrying capacity at low temperature (4°C, growth restrictive) over the course of 24hrs (the difference with the experiment presented in the main text is that inactivated phage were not removed over the course of this pilot study).
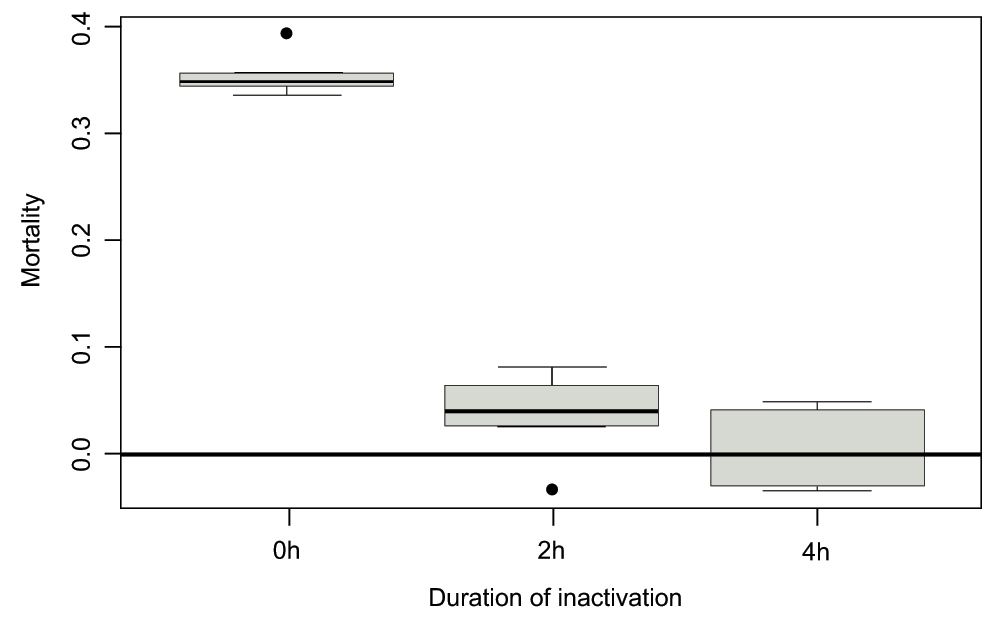
After 4 hours of exposure to UV, we observed that phages do not introduce significant mortality in the bacterial population. Kruksal-Wallis test (df = 2, p = 0.02) reveals differences between treatments, with 0h and 2h being significantly different (t-test, df = 7, p < 10-5), 0h/4h being significantly different (t-test, df = 8, p < 10-5), and 2h and 4h being similar (t-test, p = 0.16) - all p-values were Bonferroni-corrected to account for multiple testing. Similarly, 2h and 4h are not significantly different from 0 (p-values of -5 -5 0.14 and 0.89 respectively, after correction).
givegrowth = function (y, x = c(1:length(y)), bw = 12)
## y : optical density
## x : times of the measures
## bw : number of points to include in regression
{
list.of.coeff < - NULL
for (i in 1:(length(x) - bw)) {
part.x < - x[i:(i + bw)]
part.y < - y[i:(i + bw)]
cur.lm < - lm(part.y ~ part.x)$coeff[2]
list.of.coeff[i] < -cur.lm
}
result < -max(list.of.coeff)
pos < -match(max(list.of.coeff), list.of.coeff)
coeff < -lm(y[pos:(pos + bw)] ~ x[pos:(pos + bw)])$coeff
return(as.numeric(coeff[2]))
}
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)