Keywords
Metagenome-wide Association Study, Feature Extraction, Machine Learning, Rank Aggregation
This article is included in the Artificial Intelligence and Machine Learning gateway.
This article is included in the Bioinformatics gateway.
Metagenome-wide Association Study, Feature Extraction, Machine Learning, Rank Aggregation
Recent metagenomic studies of the gut microbiome have linked dysbiosis to many human diseases1–3. The identification of microbial taxa associated with human disease has been one of the important efforts in metagenomics data analysis4. Various metagenomic studies use parametric or non-parametric statistical tests to detect differentially abundant individual taxa between disease and control groups5–9. These types of methods can potentially miss taxa with weak associations but together can present strong statistical association.
In order to capture group association, several methods are proposed by exploring related taxa on a phylogenetic taxonomic tree10–14.
These elaborated statistical methods enhanced the detection of the microbial group association. However, they may still fail to detect complex multivariate nonlinear associations.
Alternative approaches of using machine learning (ML) models have been advocated for the prediction of the host phenotype15. This is motivated by the findings that a microbial signature for the host phenotype may be complex, involving simultaneous over- and under-representations of multiple microbial taxa potentially interacting with each other. Classical ML models, such as Random Forest (RF), Logistic Regression and Support Vector Machines (SVMs), and deep neural networks (DNNs) have been applied to host phenotype prediction using microbial abundance features16–22.
While the ML approaches demonstrated promising results on host phenotype prediction23,24, it is a challenging task for users to determine what is the best ML model and how many features are needed in order to achieve robust prediction, especially on external validation datasets. In addition, each ML algorithm may generate different feature importance rankings12,22,25, complicating the decision on a consistent and informative signature for the host phenotype of interest.
In this work, we introduce a novel tool, Meta-Signer, a Metagenomic Signature Identifier based on rank aggregation of informative taxa learned from individual ML models. Meta-Signer uses RF, SVM, penalized Logistic Regression, and multiple-layer perceptron neural network (MLPNN) models to evaluate importance of each microbial taxon and generates a ranked list of features (i.e., taxa) per model. It aggregates all the ranked lists using a procedure "RankAggreg" based on the cross-entropy method or the genetic algorithm26. Finally, Meta-Signer reports the top ranking features specified by the user and generates the ML models using these features. Meta-Signer’s workflow is shown in Figure 1.
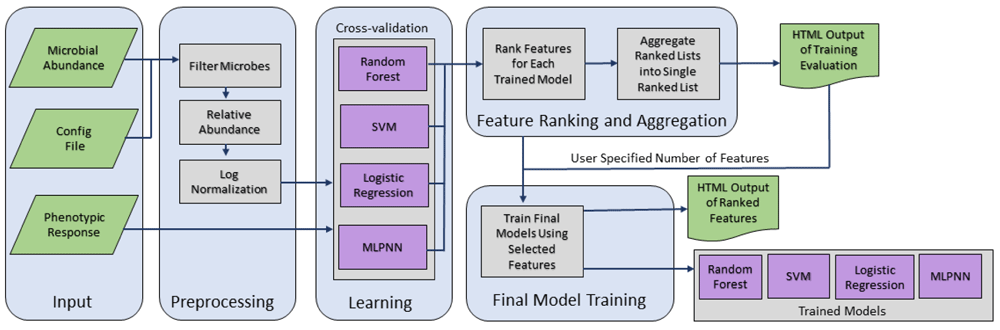
Large rounded rectangles represent different modules of the workflow. Microbial abundance is preprocessed and filtered, and then used to train ML models. Features are ranked for each model and an overall aggregated feature ranking is constructed. Meta-Signer generates portable, user-friendly HTML files for visualization as well as ML models trained on a subset of high ranking features. SVM, support Vector Machine; MLPNN, multiple-layer perceptron neural network; ML, machine learning.
Meta-Signer is user-friendly and easy to run. It provides a readable summaries as HTML outputs as well as final trained ML models. Meta-Signer is distributed as Python tool and available at https://github.com/YDaiLab/Meta-Signer.
The inputs to Meta-Signer are (1) a tab separated file of taxa abundance values where each row represents a taxon and each column represents a sample, and (2) a line separated list of response values where each row represents the phenotypic response of a sample. The first column in the abundance table should be the taxonomic identification of the taxon. An additional required file is (3) the run configuration file with user specified parameters. A final optional file is (4) the model parameters for the neural network architectures in JSON format. If this file is not found, Meta-Signer will tune the parameters and save them for later use.
Meta-Signer includes three classic ML models (RF, Linear SVM, Logistic Regression), as well as an MLPNN model. RFs are decision tree learning models trained in an ensemble fashion, taking the average of the ensemble to give a robust decision tree27. While growing each tree, a decision is made at each node by selecting the feature from a random subset of features that best splits the data into two subsets based on the Gini impurity of each subset. Given a set of data points with k classes, let pi be the proportion of samples of class i for i ∈ {1...k}. The Gini impurity of the set is calculated as
Our method implements the RF model using the scikit-learn Python library. Once trained, features are then extracted by evaluating the mean decrease impurity. For each node, the importance of the feature node being split on the decision tree is calculated as the decrease in Gini impurity before and after the split. This value is then weighted by the proportion of total samples that were split upon that node. A feature’s importance is then calculated by averaging the weighted importance values of nodes that split using that feature across all trees in the ensemble.
SVMs are supervised ML models that learn the best hyperplane to separate two classes of data28. In case of linear SVMs, a set of weights (w), and an intercept (b) will be learned. The class of the sample xi can then be determined as
Since the weights can be used to rank the importance of features, we used the linear SVMs in Meta-Signer for feature extraction. Once trained, features can be ranked using the absolute value of the learned weight parameters. SVM models are tuned with a grid search using the scikit-learn Python library.
Logistic Regression fits a logistic function to estimate the probability of binary classification; however, it can be extended to multi-class scenarios29. The model predicts the probability of a sample xi being class 1 as
where β are the weight parameters which are learned. We apply shrinkage to reduce the total number of model parameters in the final model using L1 regularization in order to penalize the absolute value of the weights, eliminating a portion of the features to create a sparse model. Given a set of samples xi (i = 1,...,n) where each sample has m features and a binary class label yi, the model minimizes the cost
where the weight parameters are penalized with the regularization parameter λ. Logistic regression models are trained using the scikit-learn Python library. Once trained, the β values are used to rank features based on their absolute value. Neural networks are consisted of multiple layers of nodes that are fully connected with edges constituting weights30. The values of a hidden layer are a linear combination of the values from the previous layer which is passed through a non-linear activation function. More explicitly, the values of a hidden layer hl is calculated as
where hl−1 are the values from the previous hidden layer, Wl are the weights connecting hl−1 to hl, bl is a bias value, and 𝜓 is a non-linear activation function. Meta-Signer uses the Rectified Linear Unit activation function for hidden layers and the softmax activation function on the output layer. The entire network is trained using a single loss function
Here ac is the predicted softmax probability of a sample’s true class c. The second term performs L2 regularization on the network weights and is penalized by λ. Networks are trained using early stopping with a validation subset of 10% of the training data.
Before training, Meta-Signer checks for a file containing network hyper-parameters for MLPNN models. If it does not find this file, Meta-Signer will use the first partition of the cross-validation to empirically determine the hyper-parameters. This is done using another cross-validation on the training set of the first partition. In addition, using the configuration file, the user can set custom parameters and even disable any of the learning models if they do not wish to incorporate it into their results. In Meta-Signer, RF, SVM, and Logistic Regression are trained using the scikit-learn python package and MLPNN models are trained using Tensorflow.
For each ML model in each cross-validated partition, Meta-Signer extracts the feature scores and uses the scores to construct a ranked feature list. RF features were scored using a method called mean decrease impurity. For each node, the importance of the feature being split upon is calculated as the decrease in Gini impurity from before and after the split. This value is then weighted by the proportion of total samples that were split upon that node. A feature’s importance is then calculated by averaging the weighted importance values of nodes that split using that feature across all trees in the ensemble. Features in Logistic Regression and SVM models were scored based on the magnitude of their coefficients in the decision functions.
The extraction of features from DNN models is a challenging task in general. We use a procedure developed in 31 to evaluate features in MLPNNs. Briefly, the MLPNN features were evaluated by calculating the cumulative weight across all layers by taking the running product of all the weight matrices in the learned networks. The product results in a matrix that has a column for each class and a row for each feature, and the value at a given index is that feature’s cumulative impact for that class. We then consider a feature’s importance as the maximum impact across classes to create a single ranked list.
For each partition of the cross-validation, we generate a single ranked list for each of ML models. Once the entirety of the cross-validated training is complete, the entire set of all ranked lists across all models is aggregated into a single top-k ranked list by minimizing the distance between the set of ranked lists and the top-k list, where k is specified by the user in the configuration file. More specifically, given a set of ranked lists {ℓ1,...,ℓm}, the top-k ranked list, , is determined as,
Here, L is the state space of top-k rankings, wi is a weight associated with ℓi, and d(θ,ℓi) is the distance between a proposed top-k ranked list, θ, and ℓi.
The aggregation is performed using the R package RankAggreg26. This package uses either a genetic algorithm or cross-entropy based approach with Markov Chain Monte Carlo sampling to find the top-k features that minimize the sum of the distances between each of the input sets and the generated top-k set. The distance used is the Spearman’s Correlation. Each input ranked list is weighted in the aggregation by the area under the receiver operating curve (AUC). After the model predictions are evaluated and the features are ranked into a single list, Meta-Signer provides a summary of the results in a portable HTML file. The file contains a description of the run and evaluation metrics for the different models in the form of boxplots. It also provides the distribution of the feature importance scores for each ML model. Lastly, it provides a list of the top-k taxa selected from the original taxa, the proportion of individual ranking sets that each taxon was present in the top-k, the rank and p-value under a PERMANOVA test32, and the class in which the taxon was found to be predictive for. All images are encoded into the file, allowing the HTML file to be moved without considering the location of the images.
Meta-Signer’s general workflow proceeds as described above and as shown in Figure 1. Meta-Signer was designed for Python 3.7 and requires the following Python packages: numpy, pandas, scipy, scikit-learn, scikit-bio, Tensorflow, matplotlib, seaborn. In addition, a working version of R version 3.0 or higher is required. Code and instructions for configuration and running can be found at https://github.com/YDaiLab/Meta-Signer.
In this section we will demonstrate how to run Meta-Signer on a dataset of patients with inflammatory bowel disease (IBD) which is provided in the GitHub repository. This dataset came from the Prospective Registry in IBD Study at MGH (PRISM)33, which enrolled patients with a diagnosis of either Crohn’s disease (CD) or ulcerative colitis (UC). The dataset includes 68 samples with CD, 53 samples with UC, and 34 healthy samples.
In addition, Meta-Signer includes another IBD dataset for external testing. This dataset consists of two independent cohorts from the Netherlands34. The first cohort consists of 22 healthy subjects who participated in the general population study LifeLines-DEEP in the northern Netherlands. The second cohort consists of subjects with IBD from the Department of Gastroenterology and Hepatology, University Medical Center Groningen, Netherlands and includes 20 samples with CD and 23 samples with UC. Together, both the PRISM dataset and the external IBD dataset included 201 microbial features. Datasets were evaluated using all three classes as well as in a binary case by combining CD and UC samples.
To use Meta-Signer and the data provided, the user needs to download Meta-Signer from the GitHub repository using the following command:
git clone https://github.com/YDaiLab/Meta-Signer.git
cd Meta-Signer
The user must make sure that all the Python and R dependencies are met before running Meta-Signer (see Operation). The user can either install these manually, or use the provided Meta-Signer Conda environment file provided using the following command:
conda env create -f meta-signer.yml
source activate meta-signer
Before running Meta-Signer, the user should open the file config.py in the src directory. This file can be used to change the run parameters of Meta-Signer and turn off and on different ML methods. An example is shown in Figure 2.

Using these parameters, we will run 10 iterations of 10-fold cross-validation on the data found in the PRISM_3 directory under the data folder. Any taxon not found in at least 10% of samples will be removed as well as any taxon with less than 0.001 mean abundance. We will use the genetic algorithm method for rank aggregation to generate a candidate list with a maximum of 50 features. Descriptions for each parameter can be found on Meta-Signer’s GitHub page as well as in the configuration file. The user can then generate the aggregated ranked list using the following command:
python generate_feature_ranking.py
This will perform the cross-validation evaluation and feature ranking, storing all the results in the results folder under a directory named after the dataset. This directory will contain internal cross-validation results as well as an HTML file that allows the user to see how different ML methods perform on the dataset. Performance of the 10 iterations of 10-fold cross-validation on the PRISM dataset is shown in Table 1. We include an analysis of both binary class (IBD vs Healthy) and tertiary class (IBD vs CD vs UC) scenarios.
Standard deviation is shown in parentheses. RF, Random Forest; SVM, Support Vector Machine; MLPNN, multiple-layer perceptron neural network; AUC, area under the receiver operating curve; MCC, Matthews correlation coefficient; F1, F1 score.
The HTML output displays these results in the form of boxplots. An example for the PRISM dataset is shown in Figure 3. In addition, Meta-Signer will evaluate the different ML methods’ training performance using increasing numbers of ranked features. Since these models will eventually overfit, the saturation of performance can help guide the user on how many features are needed to train the final model on. For the PRISM dataset, we see this saturation around 30 features, as shown in Figure 4
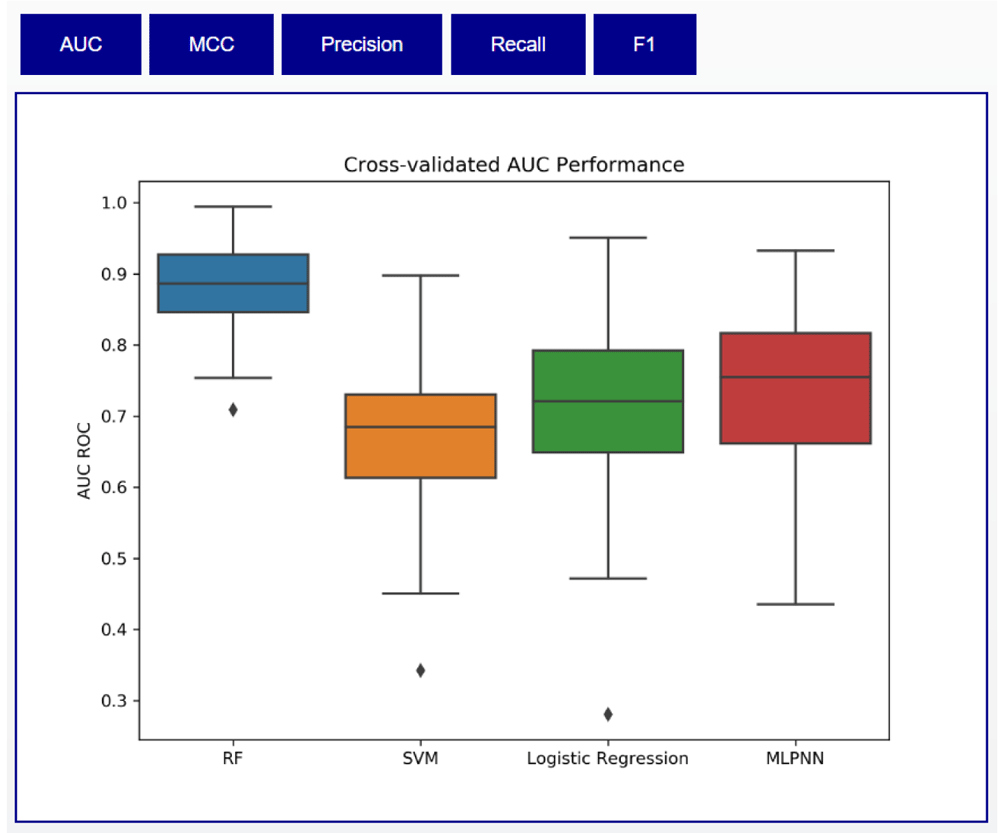
Buttons on the top allow users to cycle through different metrics. RF, Random Forest; SVM, Support Vector Machine; MLPNN, multiple-layer perceptron neural network; AUC, area under the receiver operating curve; MCC, Matthews correlation coefficient; F1, F1 score.
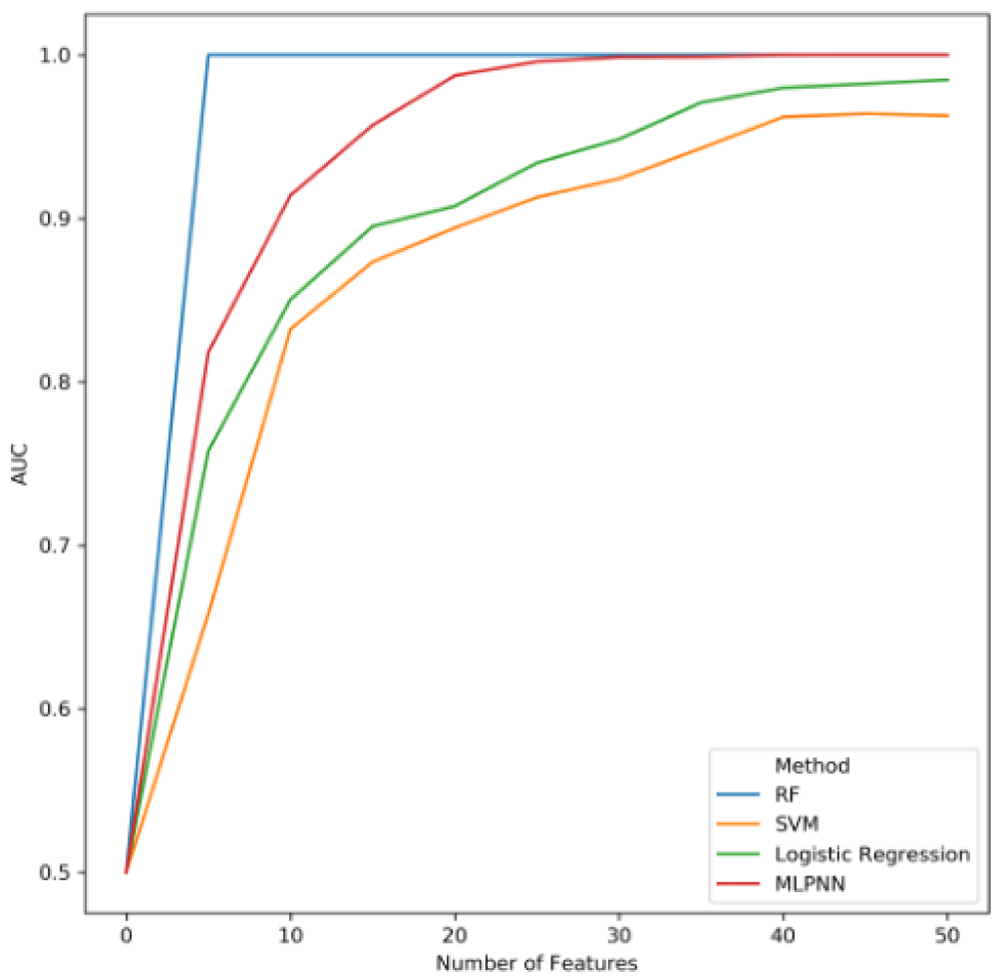
RF, Random Forest; SVM, Support Vector Machine; MLPNN, multiple-layer perceptron neural network; AUC, area under the receiver operating curve.
Using the point of saturation on the AUC curve as a guide, the user can then select the number of features and train final models using the following command:
python generate_final_models.py PRISM_3 30
Here PRISM_3 points to the result directory labeled PRISM_3 that Meta-Signer has just generated and 30 represents the number of features the user would like to have in the final models. In addition, the user can provide an external dataset to evaluate,
python generate_final_models.py
PRISM_3 30 -e PRISM_3_external
Note that PRISM_3_external must be a directory in the data directory with it’s own abundance.tsv and labels.txt files. This command will generate a directory that will contain the final trained ML models as well as another HTML file showing the ranked lists for each model as well as the aggregated ranked list as shown in Figure 5.
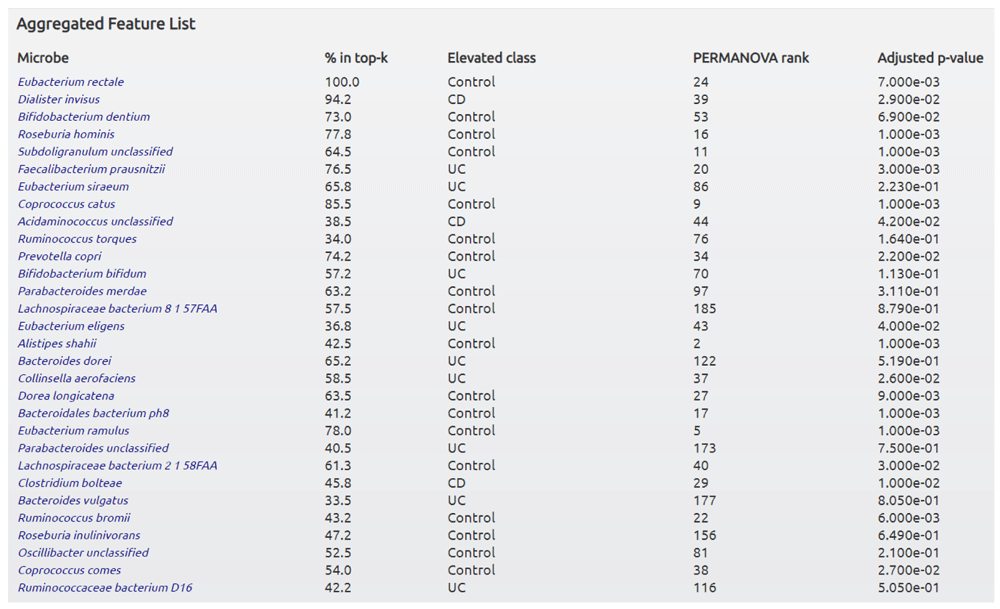
CD, Crohn’s disease; UC, ulcerative colitis.
In addition, if an external dataset is available for evaluation, the HTML file will display a table of metrics for both evaluation on the training set and evaluation on the external dataset. An example is shown in Figure 6.
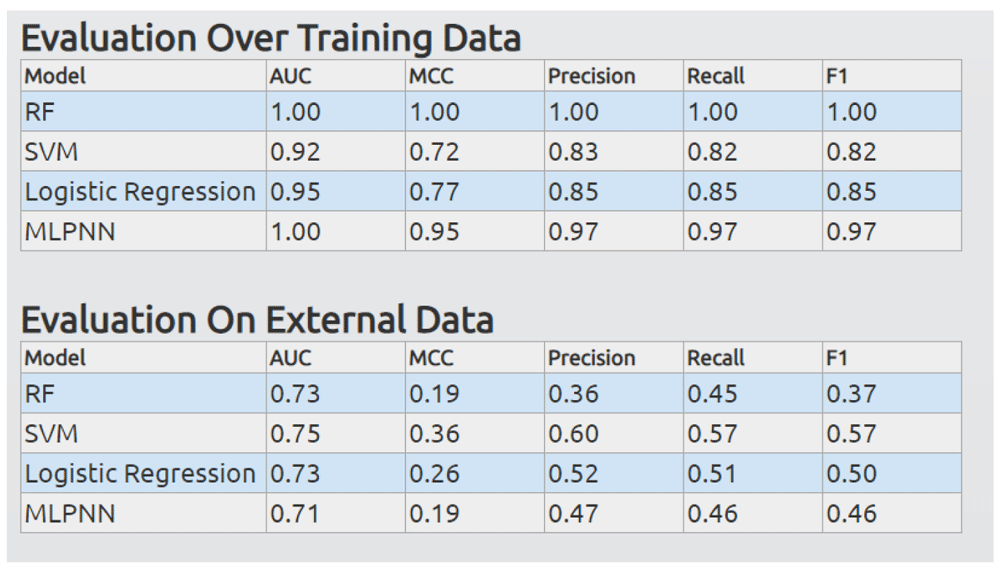
RF, Random Forest; SVM, Support Vector Machine; MLPNN, multiple-layer perceptron neural network; AUC, area under the receiver operating curve; MCC, ; F1, .
We have developed Meta-Signer, a user-friendly tool for the extraction of robust microbial taxa that are predictive to host phenotype from multiple ML models. By training different types of ML models, Meta-Signer exploits the similarities in the ranked lists of taxa learned by individual ML models to create a single aggregated set of informative microbial taxa for host phenotype prediction. We have shown that Meta-Signer can provide more informative features when compared to similar methods in both binary and multi-class scenarios.
To evaluate Meta-Signer, we benchmark against other a previously published methods Biosigner35 and a non-parametric PERMANOVA test32. Biosigner is a generic ML driven feature selection method for omics data and available in R. It uses trained RF, SVM and Partial-Least Squared Discriminant Analysis models to selectively eliminate features, resulting in a single set of remaining features. The non-parametric PERMANOVA test was included as a baseline method of feature ranking for comparison. Results for the benchmarking can be found in the Extended data document on the Meta-Signer GitHub page.
In conclusion, Meta-Signer is a user-friendly tool to identify a robust set of highly informative microbial taxa that are predictive of human disease status from a metagenomic dataset.
Original data and information for both the PRISM and external IBD datasets can be obtained from Dataset 4 of the original study http://doi.org/10.1038/s41564-018-0306-433. The data for the PRISM and external IBD datsets as formatted for use of Meta-Signer can be found at Zenodo: derekreiman/Meta-Signer: Original Release. http://doi.org/10.5281/zenodo.407740336. This project contains microbial abundance and sample class data formatted for Meta-Signer as TSV, TXT and JSON files within the folder ’data’.
Zenodo: derekreiman/Meta-Signer: Original Release. http://doi.org/10.5281/zenodo.407740336. This project contains the following extended data:
ExtendedData.pdf (Data for the benchmarking of Meta-Signer against Biosigner and PERMANOVA ranked features)
Data are under the GPL-3 license.
Source code available from: https://github.com/YDaiLab/Meta-Signer
Archived source code at time of publication: http://doi.org/10.5281/zenodo.407740336
License: GPL-3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)