Keywords
depmap, cancer, Bioconductor
This article is included in the Bioconductor gateway.
This article is included in the RPackage gateway.
depmap, cancer, Bioconductor
The consequences of genomic alterations of cancer cells on the molecular biological landscape of the cell may result in differential vulnerabilities, or “dependencies” compared to those of healthy cells. An example of genetic dependency is a gene not necessary for the survival in healthy cells, but due to perturbations of the metabolic networks caused by cancer mutations, such a gene becomes essential for the vitality of a particular cancer cell line. However, due to the complex nature of metabolic networks, the exact mechanistic nature of many genetic dependencies of cancer are not completely understood.1 A map illustrating the relationships between the genetic features of cancer and those of cancer dependencies is therefore desirable. The Cancer Dependency Map or “DepMap”, a collaborative initiative between the Broad Institute and the Wellcome Sanger Institute, aims to map genetic dependencies in a broad range of cancer cell lines. Over 1700 cancer cell lines have been selected to be tested in this effort, intended to reflect the overall distribution of various cancer diseases in the general population. The stated aim of the DepMap Project is developing a better understanding of the molecular biology of cancer and the exploiting of this knowledge to develop new therapies in precision cancer medicine.2
The DepMap initiative is, as of the date of this publication, an ongoing project, with new data releases of select datasets every 90 days. As of the 20Q4 DepMap release, 1812 human cancer cell lines have been mapped for dependencies.2 The DepMap project utilizes CRISPR gene knockout as the primary method to map genomic dependencies in cancer cell lines.2-5 The resulting genetic dependency score displayed in the DepMap data is calculated from the observed log fold change in the amount of shRNA detected in pooled cancer cell lines after gene knockout.6,7 To correct for potential off-target effects of gene knockout in overestimating dependency with CRISPR, the DepMap initiative utilized the CERES algorithm to moderate the final dependency score estimation.3 It should be noted that due to improvements in the CERES algorithm to estimate genetic dependency while accounting for CRISPR seed effects, the RNAi dependency measurements have been rendered redundant, and further data releases for RNAi dependency measurement have been discontinued as of the 19Q3 release.2,4 In addition to genomic dependency measurements of cancer cell lines, chemical dependencies were also measured by the DepMap PRISM viability screens that as of the 20Q4 release, tested 4,518 compounds against 578 cancer cell lines.2,8 A new protemic dataset was added with the 20Q2 release, providing normalized quantitative profiling of proteins of 375 cancer cell lines by mass spectrometry.9 The DepMap project has also compiled additional datasets detailing molecular biological characterization of cancer cell lines, including WES genomic copy number, Reverse Phase Protein Array (RPPA) data, TPM gene expression data for protein coding genes and genomic mutation call data. Core datasets such as CRISPR viability screens, TPM gene expression, WES copy number and genomic mutation calls are updated quarterly on a release schedule. All datasets are made publicly available under CC BY 4.0 licence.2
A table of the datasets available for the depmap package (as of 20Q4 release) is displayed in Table 1.
The ‘Release’ column indicates the most recent available release.
The depmap Bioconductor package was created in order to efficiently exploit these rich datasets and to promote reproducible research, facilitated by importing the data into the R environment. The value added by the depmap Bioconductor package includes cleaning and converting all datasets to long format tibbles,10 as well as adding the unique key depmap_id for all datasets. The addition of the the unique key depmap_id aides the comparison and benchmarking of multiple molecular features and streamlines the datasets for usage of common R packages such as dplyr11 and ggplot2.12
As new DepMap datasets are continuously released on a quarterly basis, it is not feasible to include all dataset files in binary directly within the directory of the depmap R package. To keep the package lightweight, the depmap package utilizes and fully depends on the ExperimentHub package13 to store and retrieve all versions of the DepMap data (as of this publication, starting from version 19Q1 through 20Q4) in the Cloud using AWS. The depmap package contains accessor functions to directly download and cache the most current datasets from the Cloud into the local R environment. Specific datasets (such as datasets from older releases), which can be downloaded separately, if desired. The depmap package was designed to enhance reproducible research by ensuring datasets from all releases will remain available to researchers. The depmap R package is available as part of Bioconductor at: https://bioconductor.org/packages/depmap.
Dependency scores are the features of primary interest in the DepMap Project datasets. These measurements can be found in datasets crispr and rnai, which contain information on genetic dependency, as well as the dataset drug_sensitivity, which contains information pertaining to chemical dependency. The genetic dependency can be interpreted as an expression of how vital a particular gene for a given cancer cell line. For example, a highly negative dependency score is derived from a large negative log fold change in the population of cancer cells after gene knockout or knockdown, implying that a given cell line is highly dependent on that gene in maintaining metabolic function. Genes that are not essential for non-cancerous cells but display highly negative dependency scores for cancer cell lines, may be interesting candidates for research in targeted cancer medicine. In this workflow, we will describe exploring and visualizing several DepMap datasets, including those that contain information on genetic dependency.
Below, we start by loading the packages need to run this workflow.
library("depmap")
library("ExperimentHub")
library("dplyr")
library("ggplot2")
library("stringr")
The depmap datasets are too large to be included into a typical package, therefore these data are stored in the Cloud. There are two ways to access the depmap datasets. The first such way calls on dedicated accessor functions that download, cache and load the latest available dataset into the R workspace. Examples for all available data are shown below:
rnai <- depmap_rnai() crispr <- depmap_crispr() copyNumber <- depmap_copyNumber() TPM <- depmap_RPPA() RPPA <- depmap_TPM() metadata <- depmap_metadata() mutationCalls <- depmap_mutationCalls() drug_sensitivity <- depmap_drug_sensitivity() proteomic <- depmap_proteomic()
Alternatively, a specific dataset (from any available release) can be accessed through Bioconductor’s ExperimentHub. The ExperimentHub() function creates an ExperimentHub object, which can be queried for specific terms of interest. The list of datasets available that correspond to the query, depmap are shown below:
## create ExperimentHub query object eh <- ExperimentHub() query(eh, "depmap")
## ExperimentHub with 48 records ## # snapshotDate(): 2020-10-27 ## # $dataprovider: Broad Institute ## # $species: Homo sapiens ## # $rdataclass: tibble ## # additional mcols(): taxonomyid, genome, description, ## # coordinate_1_based, maintainer, rdatadateadded, preparerclass, tags, ## # rdatapath, sourceurl, sourcetype ## # retrieve records with, e.g., 'object[["EH2260"]]' ## ## title ## EH2260 | rnai_19Q1 ## EH2261 | crispr_19Q1 ## EH2262 | copyNumber_19Q1 ## EH2263 | RPPA_19Q1 ## EH2264 | TPM_19Q1 ## ... ... ## EH5358 | crispr_21Q1 ## EH5359 | copyNumber_21Q1 ## EH5360 | TPM_21Q1 ## EH5361 | mutationCalls_21Q1 ## EH5362 | metadata_21Q1
Specific datasets can be downloaded, cached and loaded into the workspace as tibbles by selecting each dataset by their unique EH numbers. Shown below, datasets from the 20_Q3 release are downloaded in this way.
## download and cache required datasets crispr <- eh[["EH3797"]] copyNumber <- eh[["EH3798"]] TPM <- eh[["EH3799"]] mutationCalls <- eh[["EH3800"]] metadata <- eh[["EH3801"]] proteomic <- eh[["EH3459"]]
By importing the depmap data into the R environment, the data may be mined more effectively utilzing R data manipulation tools. For example, molecular dependency for all cell lines pertaining to soft tissue sarcomas, sorted by genes with the greatest dependency, can be accomplished with the following code, using functions from the dplyr package. Below, the crispr dataset is selected for cell lines with “SOFT_TISSUE” in the CCLE name, and displaying a list of the highest dependency scores.
## list of dependency scores crispr %>% dplyr::select(cell_line, gene_name, dependency) %>% dplyr::filter(stringr::str_detect(cell_line, "SOFT_TISSUE")) %>% dplyr::arrange(dependency)
## # A tibble: 815,355 x 3 ## cell_line gene_name dependency ## <chr> <chr> <dbl> ## 1 RH18DM_SOFT_TISSUE RAN -4.36 ## 2 RH18DM_SOFT_TISSUE PSMB6 -3.82 ## 3 RH18DM_SOFT_TISSUE C1orf109 -3.67 ## 4 RH30_SOFT_TISSUE RAN -3.20 ## 5 RH18DM_SOFT_TISSUE SNU13 -3.07 ## 6 RH18DM_SOFT_TISSUE SPATA5L1 -3.04 ## 7 RH18DM_SOFT_TISSUE HSPE1 -3.03 ## 8 RH18DM_SOFT_TISSUE POLR1C -2.96 ## 9 RH18DM_SOFT_TISSUE CDC16 -2.84 ## 10 RH30_SOFT_TISSUE BUB3 -2.83 ## # ... with 815,345 more rows
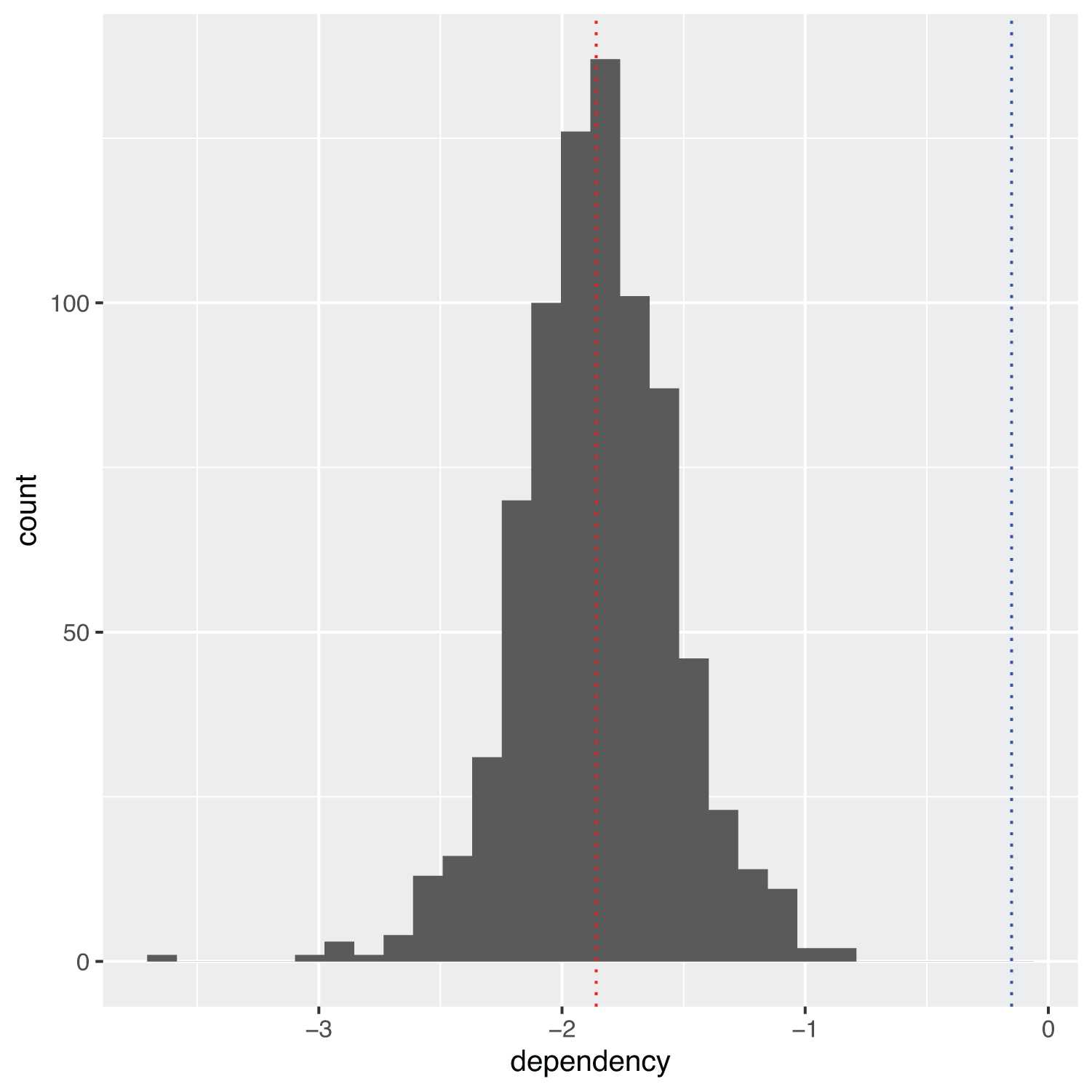
A brief survey of the top dependency scores identifies the gene C1orf109 among the most dependent genes found in the selected list of dependencies scores for soft tissue cancer cell lines. This gene, also known by the alias Chromosome 1 Open Reading Frame 109, codes for a poorly characterized protein which is theorized to promote cancer cell proliferation by controlling the G1 to S phase transition.14 This protein is selected as an interesting candidate target to explore and visualize the depmap data. Figure 1 displays the crispr data as a histogram showing the distribution of dependency scores for gene C1orf109. The red dotted line signifies the mean dependency score for that gene, while the blue dotted line signifies the global mean dependency score for all crispr measurements.
mean_crispr_dep <- crispr %>% dplyr::select(gene_name, dependency) %>% dplyr::filter(gene_name == "C1orf109") crispr %>% dplyr::select(gene, gene_name, dependency) %>% dplyr::filter(gene_name == "C1orf109") %>% ggplot(aes(x = dependency)) + geom_histogram() + geom_vline(xintercept = mean(mean_crispr_dep$dependency, na.rm = TRUE), linetype = "dotted", color = "red") + geom_vline(xintercept = mean(crispr$dependency, na.rm = TRUE), linetype = "dotted", color = "blue")
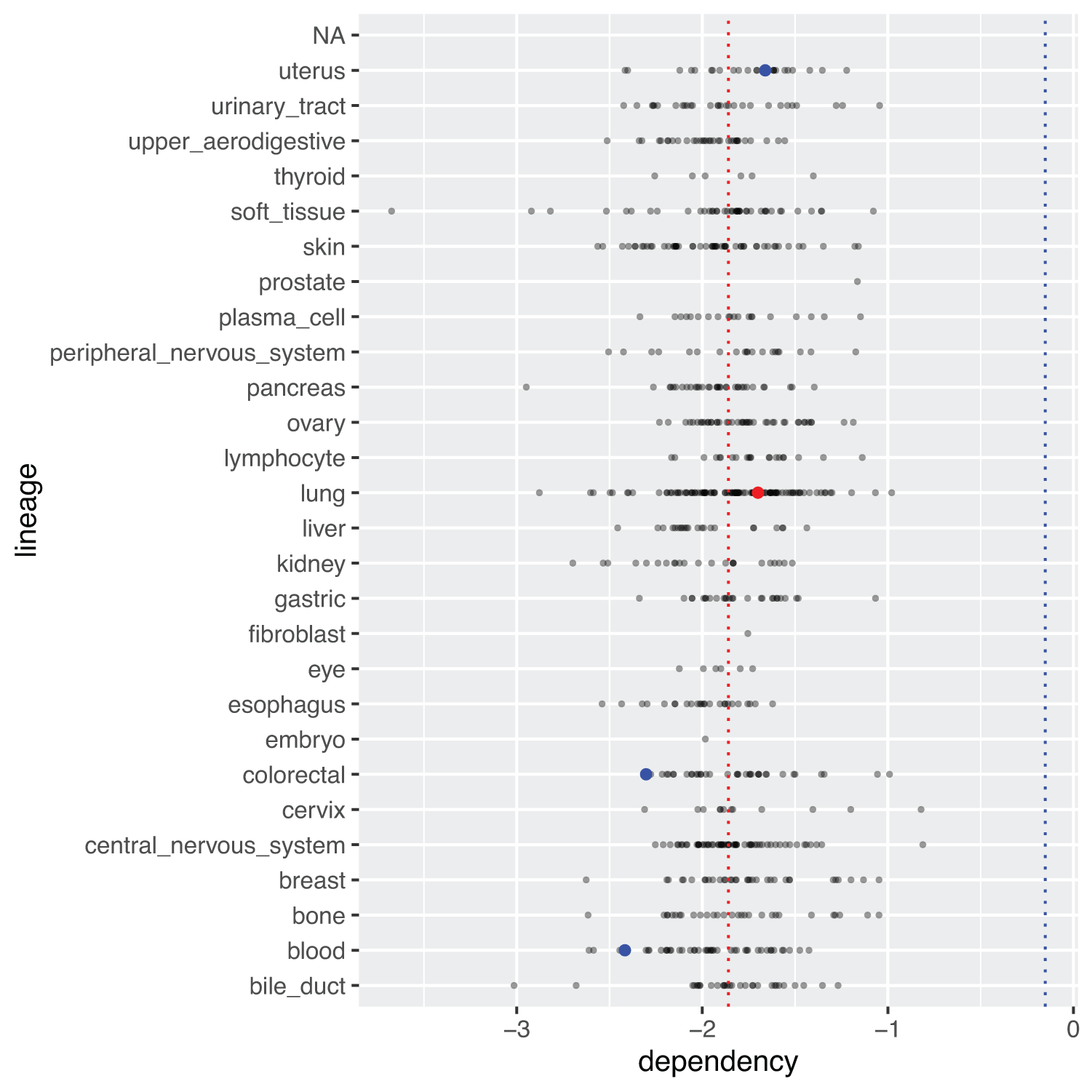
A more complex plot of the crispr dependency data, is shown in Figure 2. Visualizing this data involves plotting the distribution of dependency scores for gene C1orf109 for each major type of cancer, while highlighting the qualitative nature of mutations of this gene in such cancer cell lines (e.g. if such mutations are damaging or conserved, etc.). Genes known to be damaging mutations for a given cancer cell line are highlighted in red, while other non-conserving mutations are highlighted in blue. Notice that the plot in Figure 1 reflects the same overall distribution in two dimensions.
meta_crispr <- metadata %>%
dplyr::select(depmap_id, lineage) %>%
dplyr::full_join(crispr, by = "depmap_id") %>%
dplyr::filter(gene_name == "C1orf109") %>%
dplyr::full_join((mutationCalls %>%
dplyr::select(depmap_id, entrez_id,
is_cosmic_hotspot,
var_annotation)),
by = c("depmap_id", "entrez_id"))
meta_crispr %>%
ggplot(aes(x = dependency, y = lineage)) +
geom_point(alpha = 0.4, size = 0.5) +
geom_point(data = subset(meta_crispr,
var_annotation == "damaging"),
color = "red") +
geom_point(data = subset(meta_crispr,
var_annotation == "other non-conserving"),
color = "blue") +
geom_vline(xintercept = mean(meta_crispr$dependency, na.rm = TRUE),
linetype = "dotted", color = "red") +
geom_vline(xintercept = mean(crispr$dependency, na.rm = TRUE),
linetype = "dotted", color = "blue")
Many cancer phenotypes may be the result of changes in gene expression.15-17 The extensive coverage of the depmap data affords visualization of genetic expression patterns across many major types of cancer. Elevated expression of gene C1orf109 in lung cancer tissue has been reported in literature.14 Figure 3 below shows a boxplot illustrating expression values for gene C1orf109 by lineage:
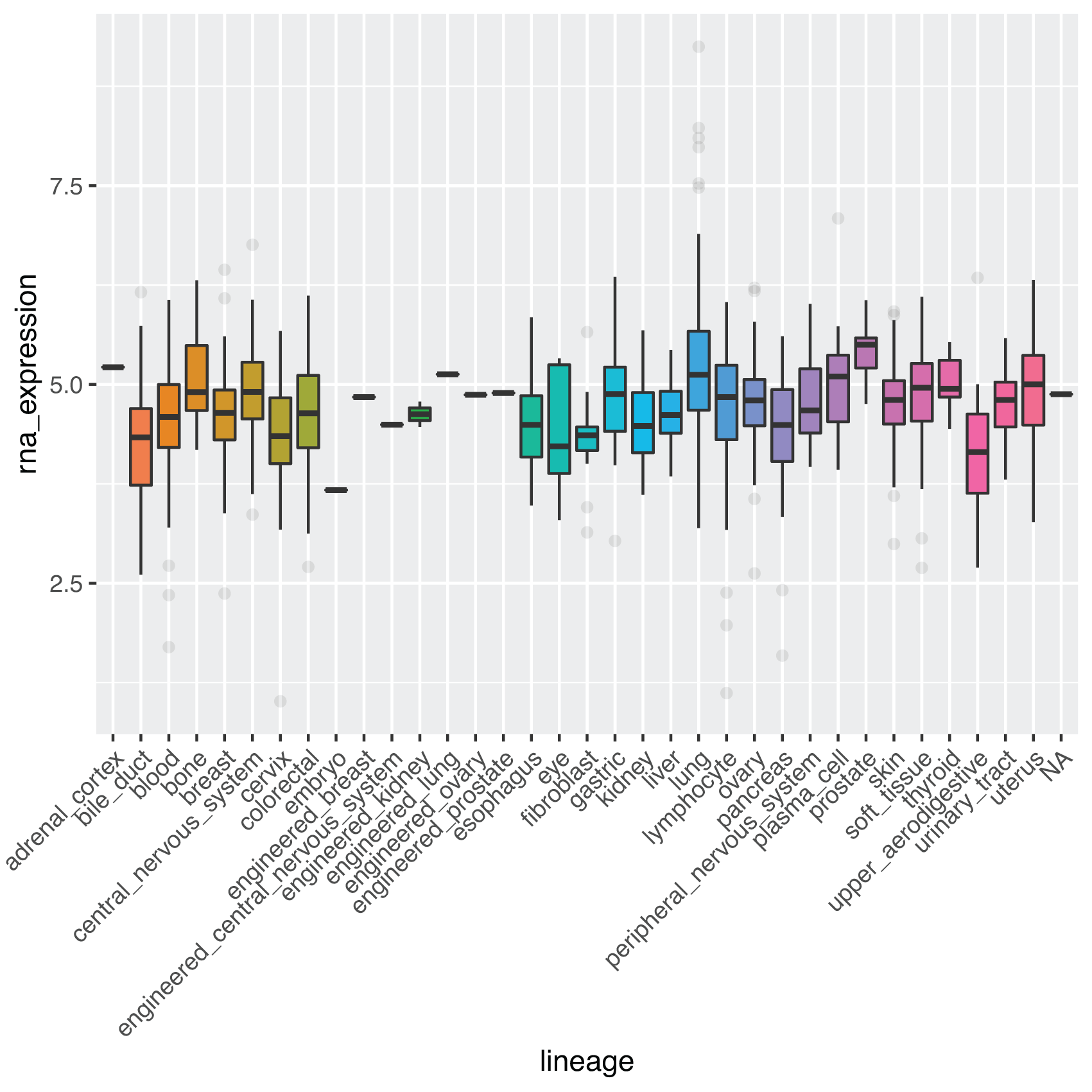
metadata %>% dplyr::select(depmap_id, lineage) %>% dplyr::full_join(TPM, by = "depmap_id") %>% dplyr::filter(gene_name == "C1orf109") %>% ggplot(aes(x = lineage, y = rna_expression, fill = lineage)) + geom_boxplot(outlier.alpha = 0.1) + theme(axis.text.x = element_text(angle = 45, hjust = 1)) + theme(legend.position = "none")
A relationship between elevated gene expression and genetic dependency in cancer cell lines has been reported in literature.1,7 Therefore, genes with elevated gene expression and high genetic dependency may present especially interesting research targets which may be explored through the DepMap datasets. Figure 4 shows a plot of expression versus CRISPR gene dependency for Rhabdomyosarcoma. The red vertical line represents the average gene expression for this form of cancer, while the horizontal line represents the average dependency for this cancer type.
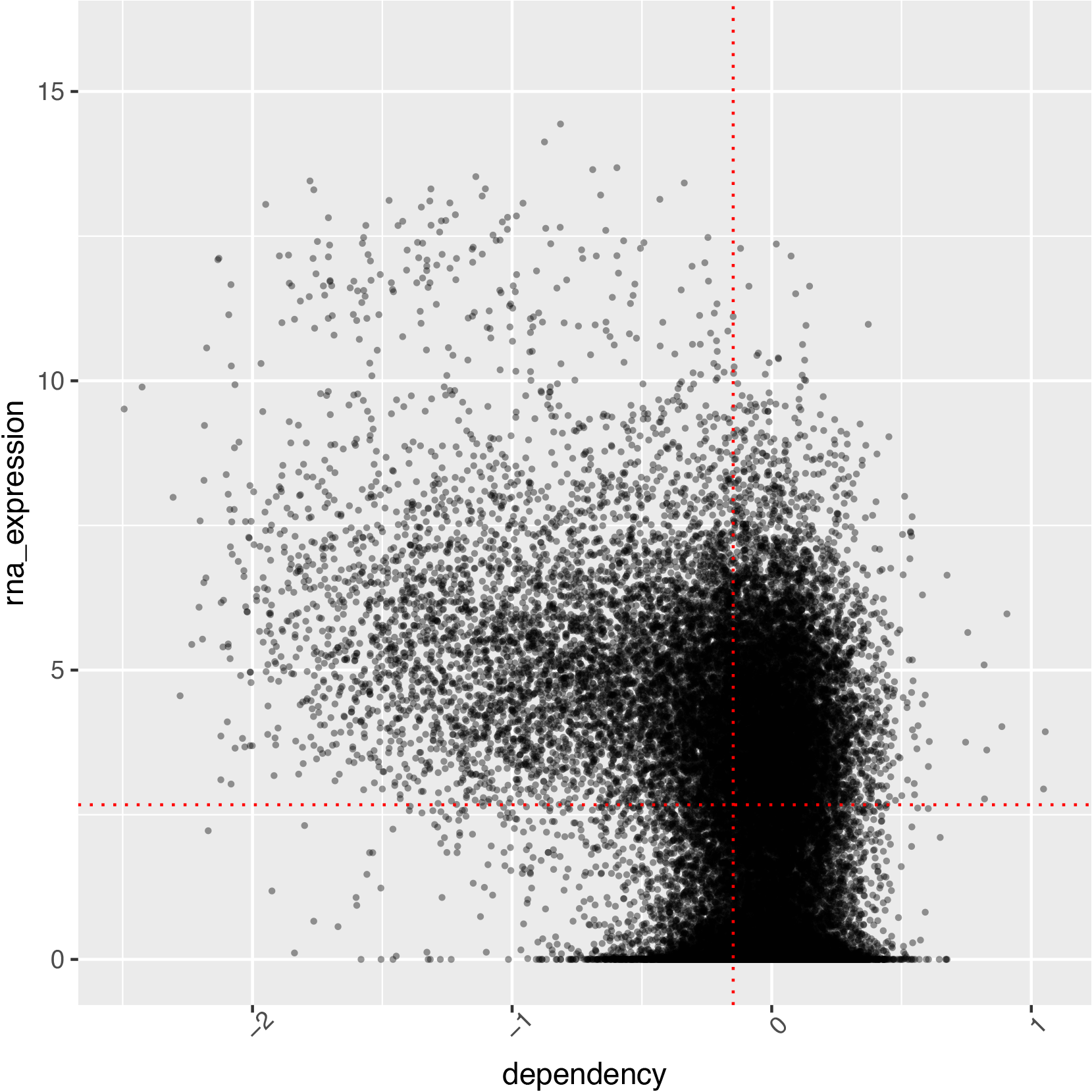
sarcoma <- metadata %>%
dplyr::select(depmap_id, cell_line, primary_disease, subtype_disease) %>%
dplyr::filter(primary_disease == "Sarcoma",
subtype_disease == "Rhabdomyosarcoma")
crispr_sub <- crispr %>%
dplyr::select(depmap_id, gene, gene_name, dependency)
tpm_sub <- TPM %>%
dplyr::select(depmap_id, gene, gene_name, rna_expression)
sarcoma_dep <- sarcoma %>%
dplyr::left_join(crispr_sub, by = "depmap_id") %>%
dplyr::select(-cell_line, -primary_disease,
-subtype_disease, -gene_name)
sarcoma_exp <- sarcoma %>%
dplyr::left_join(tpm_sub, by = "depmap_id")
sarcoma_dat_exp <- dplyr::full_join(sarcoma_dep, sarcoma_exp,
by = c("depmap_id", "gene")) %>%
dplyr::filter!is.na(rna_expression))
sarcoma_dat_exp %>% ggplot(aes(x = dependency, y = rna_expression)) + geom_point(alpha = 0.4, size = 0.5) + geom_vline(xintercept = mean(sarcoma_dat_exp$dependency, na.rm = TRUE), linetype = "dotted", color = "red") + geom_hline(yintercept = mean(sarcoma_dat_exp$rna_expression, na.rm = TRUE), linetype = "dotted", color = "red") + theme(axis.text.x = element_text(angle = 45))
Genes with the highest depenency scores and highest TPM gene expression are found in the upper left section of the plot in Figure 4. Almost all of the genes with the highest dependency scores display above average expression.
sarcoma_dat_exp %>% dplyr::select(cell_line, gene_name, dependency, rna_expression) %>% dplyr::arrange(dependency, rna_expression)
## # A tibble: 95,720 x 4 ## cell_line gene_name dependency rna_expression ## <chr> <chr> <dbl> <dbl> ## 1 JR_SOFT_TISSUE RAN -2.49 9.51 ## 2 SCMCRM2_SOFT_TISSUE RAN -2.43 9.89 ## 3 SCMCRM2_SOFT_TISSUE SNRPD1 -2.31 7.99 ## 4 JR_SOFT_TISSUE C1orf109 -2.28 4.56 ## 5 SCMCRM2_SOFT_TISSUE ATP6V1B2 -2.23 5.44 ## 6 SCMCRM2_SOFT_TISSUE POLR2L -2.21 6.09 ## 7 SCMCRM2_SOFT_TISSUE PSMA3 -2.20 7.58 ## 8 JR_SOFT_TISSUE TXNL4A -2.19 5.53 ## 9 SCMCRM2_SOFT_TISSUE POLR2I -2.19 6.51 ## 10 JR_SOFT_TISSUE SNRPD1 -2.19 8.28 ## # ... with 95,710 more rows
Evidence that changes in genomic copy number may also play a role in some cancer phenotypes has also been described in literature.3,18,19 This information can also be explored through the depmap datasets displaying the log genomic copy number across cancer lineages. Figure 5 shows such a plot for gene C1orf109 for each major type of cancer lineage:
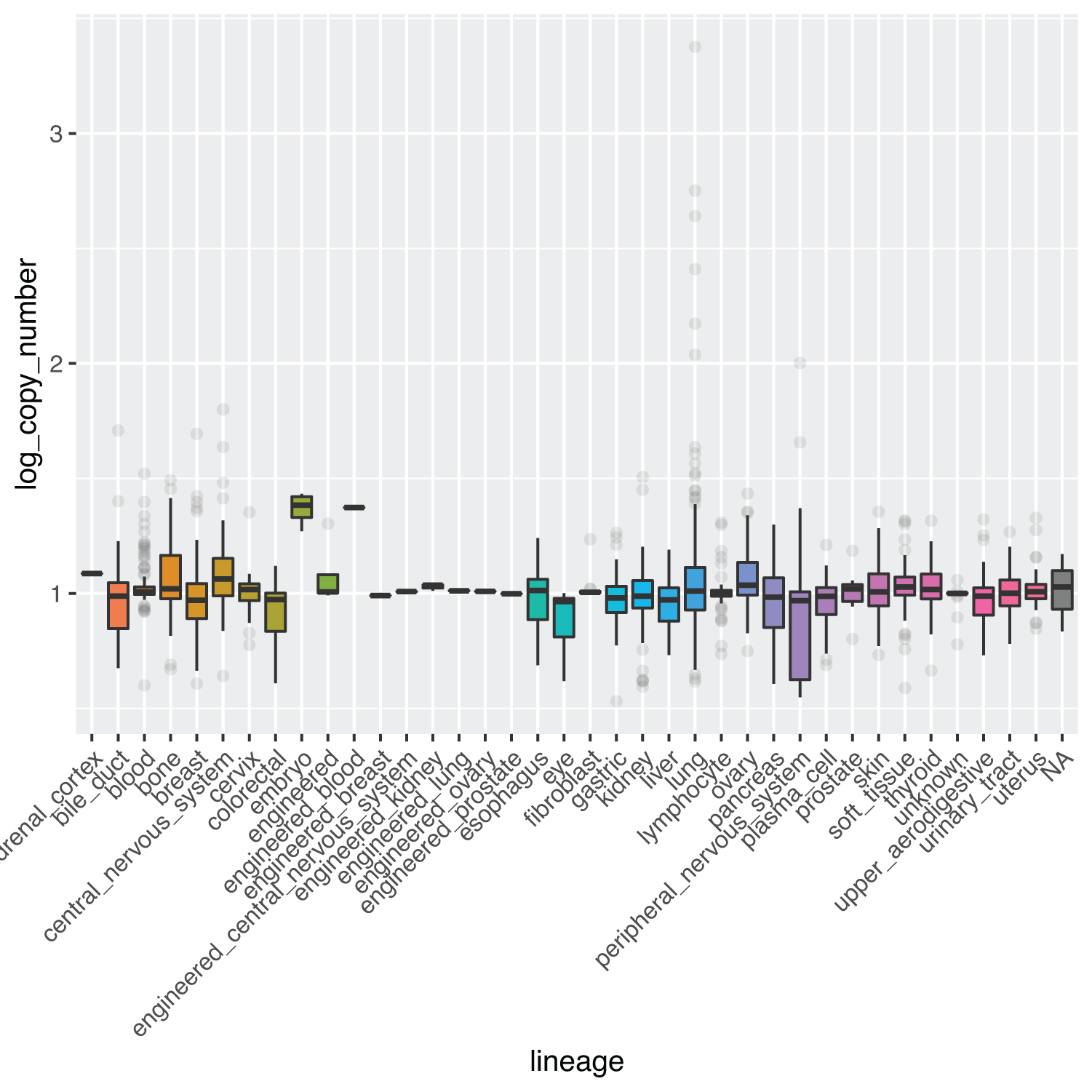
metadata %>% dplyr::select(depmap_id, lineage) %>% dplyr::full_join(copyNumber, by = "depmap_id") %>% dplyr::filter(gene_name == "C1orf109") %>% ggplot(aes(x = lineage, y = log_copy_number, fill = lineage)) + geom_boxplot(outlier.alpha = 0.1) + theme(axis.text.x = element_text(angle = 45, hjust = 1)) + theme(legend.position = "none")
We hope that this package will be used by cancer researchers to dig deeper into the DepMap data and to support their research in precision oncology and developing targeted cancer therapies. Additionally, we highly encourage current and future depmap users to combine depmap data with other datasets, such as those found through the The Cancer Genome Atlas (TCGA) and the Cancer Cell Line Encyclopedia (CCLE).
The depmap R package will continue to be maintained in line with the biannual Bioconductor release schedule, in addition to quarterly releases of DepMap data.
We welcome feedback and questions from the community. We also highly appreciate contributions to the code in the form of pull requests on github.
The depmap datasets are available through ExperimentHub. To install the depmap package, start a recent version of R and execute:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("depmap")
The depmap package is available from: https://doi.org/doi:10.18129/B9.bioc.depmap Source code available from: https://github.com/UCLouvain-CBIO/depmap Archived source code as at time of publication: http://doi.org/10.5281/zenodo.473994920 License: Artistic-2.
All packages used in this workflow are available from the Comprehensive R Archive Network (https://cran.r-project.org) or Bioconductor (http://bioconductor.org). The specific version numbers of R and the packages used are shown below.
## R version 4.0.3 Patched (2021-01-18 r79847) ## Platform: x86_64-pc-linux-gnu (64-bit) ## Running under: Manjaro Linux ## ## Matrix products: default ## BLAS: /usr/lib/libblas.so.3.9.0 ## LAPACK: /usr/lib/liblapack.so.3.9.0 ## ## locale: ## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C ## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8 ## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 ## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C ## [9] LC_ADDRESS=C LC_TELEPHONE=C ## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C ## ## attached base packages: ## [1] parallel stats graphics grDevices utils datasets methods ## [8] base ## ## other attached packages: ## [1] stringr_1.4.0 ggplot2_3.3.3 ExperimentHub_1.16.0 ## [4] AnnotationHub_2.22.0 BiocFileCache_1.14.0 dbplyr_2.1.1 ## [7] BiocGenerics_0.36.0 depmap_1.4.0 dplyr_1.0.5 ## [10] kableExtra_1.3.4 ## ## loaded via a namespace (and not attached): ## [1] Biobase_2.50.0 httr_1.4.2 ## [3] bit64_4.0.5 viridisLite_0.3.0 ## [5] shiny_1.6.0 assertthat_0.2.1 ## [7] interactiveDisplayBase_1.28.0 BiocManager_1.30.12 ## [9] stats4_4.0.3 blob_1.2.1 ## [11] yaml_2.2.1 BiocWorkflowTools_1.16.0 ## [13] BiocVersion_3.12.0 pillar_1.5.1 ## [15] RSQLite_2.2.5 glue_1.4.2 ## [17] digest_0.6.27 promises_1.2.0.1 ## [19] rvest_1.0.0 colorspace_2.0-0 ## [21] htmltools_0.5.1.1 httpuv_1.5.5 ## [23] pkgconfig_2.0.3 bookdown_0.21.6 ## [25] purrr_0.3.4 xtable_1.8-4 ## [27] scales_1.1.1 webshot_0.5.2 ## [29] svglite_2.0.0 later_1.1.0.1 ## [31] git2r_0.28.0 tibble_3.1.0 ## [33] farver_2.1.0 generics_0.1.0 ## [35] IRanges_2.24.1 usethis_2.0.1 ## [37] ellipsis_0.3.1 cachem_1.0.4 ## [39] withr_2.4.1 cli_2.4.0 ## [41] magrittr_2.0.1 crayon_1.4.1 ## [43] mime_0.10 ps_1.6.0 ## [45] memoise_2.0.0 evaluate_0.14 ## [47] fs_1.5.0 fansi_0.4.2 ## [49] xml2_1.3.2 tools_4.0.3 ## [51] lifecycle_1.0.0 S4Vectors_0.28.1 ## [53] munsell_0.5.0 AnnotationDbi_1.52.0 ## [55] compiler_4.0.3 systemfonts_1.0.1 ## [57] rlang_0.4.10 grid_4.0.3 ## [59] rstudioapi_0.13 rappdirs_0.3.3 ## [61] labeling_0.4.2 rmarkdown_2.7 ## [63] gtable_0.3.0 DBI_1.1.1 ## [65] curl_4.3 R6_2.5.0 ## [67] knitr_1.31.3 fastmap_1.1.0 ## [69] bit_4.0.4 utf8_1.2.1 ## [71] stringi_1.5.3 Rcpp_1.0.6 ## [73] vctrs_0.3.7 tidyselect_1.1.0 ## [75] xfun_0.22
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)