Keywords
driver gene, cancer, module identification, pathway analysis, network biology, topology, reach metric, signaling network
This article is included in the Bioinformatics gateway.
driver gene, cancer, module identification, pathway analysis, network biology, topology, reach metric, signaling network
Cancer is a disease of uncontrolled cell proliferation. Genetic mutations alter operations inside normal cells in ways that promote tumorigenesis. Receptors at the cell surface gather the signals from other cells, and funnel them into the cell. Signals are transmitted from upstream proteins and passed to downstream effector proteins. These mappings are represented in signaling networks, one of which was provided as a weighted directed graph by the DREAM Challenge. Dysregulation of these pathways by driver mutations is often found in the development and progression of cancer. Known driver genes, as listed in the Cancer Genome Census (CGC)1, within this signaling network provided points of reference. Disturbance to these driver genes or their upstream parents by mutations in either parent or child, can lead to disruption of highly regulated signals affecting the normal functions of cells. We explored these parents as potential candidate driver genes. We also considered driver gene parent protein products that were functionally related in physical protein-protein interaction (PPI) networks and co-expression networks as candidate driver genes.
Module or community detection is a classical problem in social and computer network science2–4. Different methods yield communities that are either too large or too small to be easily understood. Biologically relevant modules and their relevance to a disease are poorly understood5. With the increased availability of biological network data, scientists are no longer solely relying on fixed pathways6,7. They are now seeking to expand on known pathways and discover novel pathways in the analysis of diseases. The challenge assessed our modules using a number of genome-wide association studies (GWAS). Each of our modules were scored for enrichment against 104 GWAS, using their PASCAL tool8. We then classified these novel disease pathways with CGC known driver genes as cancer modules. The signaling parent genes, which formed part of these novel cancer pathways, were nominated as candidate driver genes.
The co-expression and physical Protein-Protein Interaction Network provided in Subchallenge 2 were combined. The networks were maintained as interaction lists, which proved to be more efficient than the memory demanding matrix manipulation. The network combination method employed was based on the system developed previously9. This network combination process considered the interaction existence and weighting in each of the individual networks before accepting it as part of the combined network. Linear regression was used in the calculation of the weighting in the combined network.
The individual networks are summarized in Table 1. These anonymized networks prevented any bias in the module identification process. Edges in the combined network were weighted and during the leaderboard stages this weight and its cutoff values were optimized to construct a final combined network with high-scoring edges. The clustering technique was employed on this combined network, while the signaling network was used to determine the parent-child relationships.
The number of nodes accessible within two steps as a proportion of the total nodes in the network, is defined as the 2-reach of that node. This 2-reach value was calculated for each node in the combined network. By repeatedly selecting two genes with the highest 2-reach values from the set of genes not yet assigned to a cluster, the complete network was decomposed. These two genes were the center of two separate clusters and their immediate neighbors chosen as members. To prevent overlapping, clusters were removed from the network, resulting in further fragmentation; such fragments give rise to clusters themselves as they may be within a range of 3–100 members. The largest remaining fragment was decomposed by repeating the 2-reach cluster formation process.
The DREAM Challenge assessed modules based on a collection of GWAS, which is superior to matching the predicted modules against well-known pathway databases or annotated information. The unique GWAS used in the assessment was not part of the construction of the networks provided. The Challenge used these GWAS and our modules as input to the PASCAL scoring tool8, which provided 104 p-values for each of our modules in the final submission. With these multiple p-values, for each of our modules, we were then required to perform a level of correction to control the False Discovery Rate (FDR) using the Benjamini-Hochberg procedure10, as some of the p-values less than 0.05 may have been by chance.
Significant modules (SM) with at least one driver gene from the CGC list were classified as cancer modules (CM). These cancer modules were a subset of the set of significant disease modules as depicted in Figure 1A. The list of genes in these cancer modules (CMgenes), were a source of potential driver genes. Additionally, from the signaling network in Table 1, we identified a list of parent genes (Pgenes) to well-known driver genes as another source of potential driver genes. Figure 1B highlights the intersection of these two lists. The CMgenes were based on the network module identification and GWAS scoring, while the Pgenes were based on the parent-child relationships with well-known driver genes in the signaling network. The overlap of these two lists provided the list of candidate driver genes based on a consensus.

(A) The set of significant modules (SM) with well-known driver genes from the Cancer Genome Census list were identified as cancer modules (CM). (B) The intersection of the genes from the cancer modules CMgenes and the parents from the signaling network Pgenes highlight our first list of candidate driver genes.
The scripts and source code used to perform the analysis in this study are available as Extended data11.
The network formed by combing the co-expression network and the protein-protein interaction (PPI) network consisted of 14,200 genes and 824,528 edges. The removal of low-confidence edges resulted in the loss of 1,583 genes, 199 of which were unique to the co-expression network, 1,201 of which were unique to the PPI network, and 183 of which were common to both. The overlap of these two DREAM Challenge networks and the combined network is illustrated in Figure 2A.

(A) Genes from Combined Network and the individual networks. (B) Genes from the signaling network, Cancer Genome Census (CGC) and their parents. PPI, protein-protein interaction.
The signaling network included 5,254 nodes and 21,826 edges. Of the 616 CGC driver genes 470 were present in this network. These driver genes were children of 1,721 parents, with TP53 having 154 (the highest number of parents); 63 other driver genes were orphans. All of the orphan driver genes were themselves parents in this signaling network. Figure 2B shows the overlap of the signaling network, the CGC driver genes and their parents. We also see that 306 of the CGC driver genes are both parents and children of other driver genes.
Our 2-reach clustering algorithm produced 237 non-overlapping modules, ranging in size from 3 to 100, as shown in Figure 3A. Many of our modules were 50 genes in size, as this was the default module size in the cluster formation process. Our method also produced modules that were at the upper bound of 100 genes and the lower bound of 3 genes in size, a result of fragmentation of the network. The 237 modules extracted from the combined network included 4,682 genes, which was less than 33% of the total network, since only the largest fragment was further assessed for clusters.
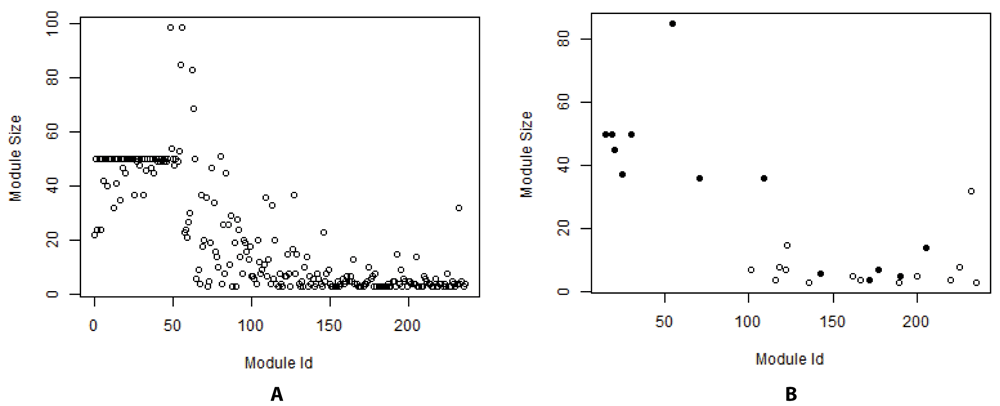
(A) All 237 modules. (B) 27 Significant modules with 13 cancer modules identified by solid circles.
On examination of the 104 multiple p-value scores provided by the DREAM Challenge for each of our modules, we found 27 significant modules existed, all shown in Figure 3B. The 13 solid circles are the CM, which contain known driver genes. It is difficult to get highly significant enrichment p-values for modules of a small size. Consider a module of size 4 that includes two significant genes. Even though 50% is certainly a clear enrichment, this could also occur by chance. In contrast, a module of size 100 that contains 50% significant genes would get an extremely small p-value. Of the 237 modules identified from our 2-reach method on the combine network, 27 were identified as significant at the 10% FDR, and 13 were cancer related as seen in Figure 3. There were different assessment stages during the challenge. Our best result was at the 2.5% FDR, where we were the fifth-best clustering method as identified in the Supplementary Figure 5 in the main paper12. Figure 4 shows the details with the performance of every method from the teams listed on the x-axis as compared to the highest scoring method from subchallenge 1. The first three teams with Bayes factor K less than three indicated their methods were better than this reference method.

The y-axis represents the number of significant (NS) modules identified in the final phase of subchallenge 2.
The 35 driver genes in these 13 cancer modules contained 84 unique parents from the signaling network. In total, 27 of the 84 parents were also driver genes, which left 57 as candidate driver genes. Many of the driver genes were also parents, but module 20, module 55, module 109, and module 143 contained driver genes that were not parents, since the “#Drivers in module” column value was larger than the number of red genes provided in Table 2. In the case of module 55, with as many as nine known driver genes, only five were parents in the signaling network. Module 143 has a unique condition of containing the single CGC driver gene HLA-A, which was not a parent, and therefore does not appear in Table 2. The presence of this driver gene, qualified module 143 to be a cancer module, but only the parents of other known driver genes were considered as candidate drivers. Each of the cancer modules CM and the candidate driver genes they contain are listed in Table 2; candidate genes are listed in black.
Additional analysis of the cancer modules further prioritized our initial list; we assessed the specific parent child relationships in the subgraphs using the parent genes for each of the modules in the signaling network. On examination of the 13 subgraphs, only three (modules 15, 19 and 55) had direct parent-child relationships, where the parent was listed in the module and the child was a CGC well known driver gene. In the case of module 15, there were five driver genes FBXW7, THRAP3, ABL1, FANCD2 and YWHAE, with 13 parent genes. The five driver genes were themselves parent genes, so we were left with eight parents to consider as potential candidate driver genes as listed in Table 2. Of the five driver genes in this module, only ABL1 had a parent from this same module in the signaling network – PRKDC, as seen in Figure 5. Similarly, module 19 showed the RAF1 driver gene had three parents in the signaling network, CDK4, YWHAB, and GSK3B. Module 55 has nine driver genes, but only five were parents. RB1 was the only driver gene with the neighboring parent PPP1CB from the signaling network. This process prioritized five parents, which are underlined in Table 2. They are neighbors of existing drivers in the cancer modules, and are parents to the same driver gene in the signaling network. Of these five, CDK4 is already present in the CGC list of known driver genes. The other four genes PRKDC, YWHAB, GSK3B, and PPP1CB are shortlisted.

Five blue nodes are the known drivers. Only driver gene ABL1 is seen to have a parent which forms part of module 15.
We used three diverse genomic networks to reveal novel candidate genes from pathways underlying cancer: the physical PPI, the functional co-expression, and the signaling networks. Our method incorporated the simple m-reach topology metric in disease module identification. The reach measure has been shown to be useful in the key player problem13, and has been used to identify cancer driver genes14. Recent publications in the literature highlight the four candidate genes that were shortlisted. PRKDC has been shown to be associated with poor clinical outcome in gastric cancer patients15. Recent studies proposed the YWHA family is able to regulate a vast number of proteins involved in key cellular processes, with implications for tumorigenesis and cancer progression16. GSK3B is known to regulate epithelial-mesenchymal transition and cancer stem cell properties, and is a novel drug target for triple-negative breast cancer17. PPP1CB has been identified as a protein safeguarding nuclear integrity; altered nuclear shape is a defining feature of cancer cells18.
Zenodo: Parent-child signals identify candidate cancer driver genes. http://doi.org/10.5281/zenodo.374080511.
This project contains the scripts and source code used to perform the analysis in this study.
Extended data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)