Keywords
Diabetes, Cardiovascular diseases, Oxidative stress, Inflammation, Non-coding RNAs, Lifestyle
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Oxidative Stress in Diabetic Complications collection.
Diabetes, Cardiovascular diseases, Oxidative stress, Inflammation, Non-coding RNAs, Lifestyle
In this version, the reviewers want to change the title of the manuscript. In addition, we change the introduction because needs to explain the type 2 diabetes. Moreover, we described long non-coding RNAs and related them to type 2 diabetes. It should be noted that we rewrote the introduction section and revised the sub-titles. Based on the comments, we attempt to improve the structure and consistency of the texts.
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
3′UTR: 3′ untranslated region
AGEs: advanced glycation end products
BAX: BCL2 Associated X
Bcl-2: B-cell lymphoma 2
BCAAs: branched-chain amino acids
CAT: catalase
Cycs: Cytochrome C
circRNAs: circular RNAs
DAMPs: damage-associated molecular patterns
DNA: deoxyribonucleic acid
Fas: Fas Cell Surface Death Receptor
FNDC5: fibronectin type III domain containing 5
GAS5: growth arrest-specific 5
GPX: Glutathione peroxidase
H2O2: hydrogen peroxide
HOTAIR: HOX transcript antisense RNA
IL-1: interleukin-1
IL-6: interleukin-6
IL-8: interleukin-8
IL-18: interleukin-18
lncRNAs: long non-coding RNAs
MAPK: mitogen-activated protein kinase
mPTP: mitochondrial permeability transition
MEG3: maternally expressed 3
NCCD: Nomenclature Committee on Cell Death
NO: nitric oxide
NCDs: non-communicable diseases
NF-κB: nuclear factor kappa B
NLRs: nucleotide binding and oligomerization domain
O2-: superoxide
OH: hydroxyl radical
PKR: protein kinases R
RIP3: receptor-interacting protein 3
ROS: reactive oxygen species
Sirt3: Sirtuin 3
SODs: superoxide dismutase
T2D: type 2 diabetes
TNF-α: tumor necrosis factor-alpha
TLR4: toll-like receptor 4
TUG1: taurine up-regulated 1
VDAC1: voltage-dependent anion channel 1
Type 2 diabetes (T2D) has been classified as one of the four primary non-communicable diseases (NCDs) that demands immediate medical attention to manage its prevalence and related problems.1 The growing evidence indicated that T2D could be a severe metabolic condition affecting more than 400 million individuals globally, and predicted this number might surpass 600 million by 2040.2,3 The statistical evidence demonstrated that T2D is a top 10 cause of death globally, killing over 1.5 million people worldwide.4 T2D is recognized by β-cell dysfunction and hyperglycemia, which result in defective or inadequate insulin receptor activity, impaired function, and early destruction of insulin during its synthesis.5,6 Immense studies have indicated that type 2 diabetes leads to severe chronic complications such as cardiovascular disease.2,7,8
Moreover, T2D is the third most significant risk factor for worldwide premature mortality due to inflammation, hyperglycemia, and oxidative stress.9 Immense studies have revealed that factors like inflammation and oxidative stress can stimulate the epigenetic regulator related to several diseases. A significant proportion of the human genome undergoes transcription into non-translated RNA molecules.10 There is a widespread recognition that these non-coding RNAs (ncRNAs) have a central role in human physiology and pathology.11 Nevertheless, in this review, we have focused on the function of the long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) in diabetes pathomechanisms as well as diabetic cardiovascular complications. The latest research has suggested that ncRNAs, which include miRNAs with a length of 19 to 25 bp, lncRNAs with a length of over 200 bp, and ring RNAs (circRNAs) comprise more than 98% of human genome expression products.12 In addition, microRNAs can bind to messenger RNAs (mRNAs) through the complementary 3′-untranslated region (3′UTR), thus effectively regulating the expression of targeted genes. It follows that miRNAs are indirectly involved in regulating multiple physiological processes. LncRNAs are characterized by non-random short open reading frames (sORFs) and play a pivotal role in fundamental biological processes. The significance of ncRNAs in maintaining the body's normal functioning has been established. Abnormal ncRNAs expression is strongly linked to several diseases, including diabetes mellitus (DM).13–15 Growing studies have emphasized ncRNAs in DM and its complications, proposing that ncRNAs can interact with insulin.16 Furthermore, there is evidence indicating that ncRNAs might act as diagnostic markers and modulators of diabetic cardiovascular disease.17–20 Impaired glucose tolerance in T2D is associated with insulin resistance, and insulin shortage impacts glucose consumption in the liver, adipose tissues, and skeletal muscle.21
Recent studies have revealed oxidative stress as a critical player in developing diabetes complications.22 Hyperglycemia causes increased oxidative stress, pro-inflammatory proteins, and infiltrating macrophages secreting inflammatory cytokines that impair the body's function.1 Moreover, It has been discovered that insulin resistance is associated with increased inflammation molecules such as tumor necrosis factor-alpha (TNF-α) production.23,24
Vascular complications, macrovascular and microvascular, are the leading causes of morbidity and mortality in people with diabetes, placing a significant economic burden on developed and developing nations due to disparities in health care spending and the inaccessibility of effective medicines.8,9 There is a well-established clinical connection between diabetes and atherosclerotic lesions.25 Foam cell production, fatty streak development, and plaque rupture are all proven to occur during atherosclerotic damage. In atherosclerotic lesions, vulnerable plaque development generates systemic inflammation, resulting in myocardial infarction.26
The release of bioactive metabolites from adipocytes, such as lipids, free fatty acids, monocyte chemoattractant protein-1, and pro-inflammatory cytokines, have been associated with insulin resistance in obese people.27,28 On the other hand, evidence has indicated that the human body has an antioxidant defense mechanism that drastically lowers the rate at which oxidizing chemicals are oxidized.29 There are both enzymatic and non-enzymatic antioxidants in the human body. Glutathione peroxidase (GPx), catalase (CAT), and superoxide dismutase (SODs) are examples of enzymes that neutralize free radicals30 (Figure 1).
Even though many pathophysiological changes are produced by metabolic disorders such as insulin resistance, hyperglycemia, and dyslipidemia, oxidative stress may have a vital role in the physiological and pathological mechanisms of T2D.31 However, immense evidence has indicated that the vital role of oxidative stress in T2D, the molecular mechanism of oxidative stress is not elucidated. Along with the development of cardiac glucotoxicity, hyperglycemia produces a rise in the concentration of ROS and the production of advanced glycation end products (AGEs). In addition, cell death is induced by ROS, which can have significant consequences that modulate pathogenic reactions in the rest of the cells, resulting in dysfunction and remodeling.32,33
Besides, oxidative stress can trigger inflammation, cytokine storm, nitric oxide (NO) accumulation, mitochondrial dysfunction, and reduced mitochondrial biogenesis.6 Furthermore, epidemiology studies demonstrated that a sedentary lifestyle and excessive sitting caused dysmetabolic conditions.34 Furthermore, a lifestyle change, including being physically active, avoiding smoking and drinking, and eating healthy, could prevent many occurrences of T2D.35,36
Based on the data mining, oxidative stress plays a key role in T2D, but the precise mechanism and essential factors related to oxidative stress are not comprehended. Hence, the main goal of this review article attempts to find out:
• The cellular mechanisms relating oxidative stress and cell death in T2D.
• The role of the non-coding RNAs and oxidative stress in managing T2D.
• Finally, we will discuss how lifestyle can enhance oxidative stress in the body.
Free radicals and other non-radical reactive derivatives are referred to as oxidants and are collectively referred to as ROS and reactive nitrogen species (RNS).37 Highly reactive molecules created during normal cellular metabolism react with various organic substrates, including lipids, deoxyribonucleic acid (DNA), and proteins.38,39 Oxidative radicals, including superoxide (O2−), hydroxyl radical (·OH), and hydrogen peroxide (H2O2), are produced at high concentrations in response to oxidative stress, leading to damaged cells and even cell death.28 Oxidative stress and cell death are hallmarks of diabetes and cardiovascular diseases, although managing these hallmarks may help postpone the development of diabetes and diabetic vascular complication.14
Oxidative stress contributes to various pathological disorders, including cancer, rheumatoid arthritis, and diabetes mellitus.40 Evidence has indicated that oxidative stress may mediate and progress T2D.41–43 Increasing the ROS concentration changes the SOD, CAT, and GPx capacity. Hence, alternation in the level of SOD, CAT, and GPx and decreased antioxidant defense lead to enhance the ROS and progression of T2D.44
Growing studies have demonstrated a correlation between oxidative stress and several biomarkers, such as DNA damage and lipid peroxidation.45 Free radicals play a significant role in the initiation and advancement of late diabetes complications, as they can inflict damage on proteins, DNA, and lipids.44,46 Deprivation stress and free radicals trigger DM, including coronary artery disease, retinopathy, neuropathy, and nephropathy.47 Moreover, hyperglycemia in the initiation of oxidative stress impairs endothelial function within the blood vessels of individuals with diabetes.44 Diabetic hyperglycemia leads to oxidative degradation and protein glycation by forming free radicals.44 The quantification of protein glycation is determined by utilizing biomarkers, including fructosamine levels and glycated hemoglobin. The modification in the function and structure of antioxidant protein enzymes can also occur due to non-enzymatic glycation, leading to an impact on the detoxification of free radicals and an increase in oxidative stress in individuals with diabetes.43,48,49
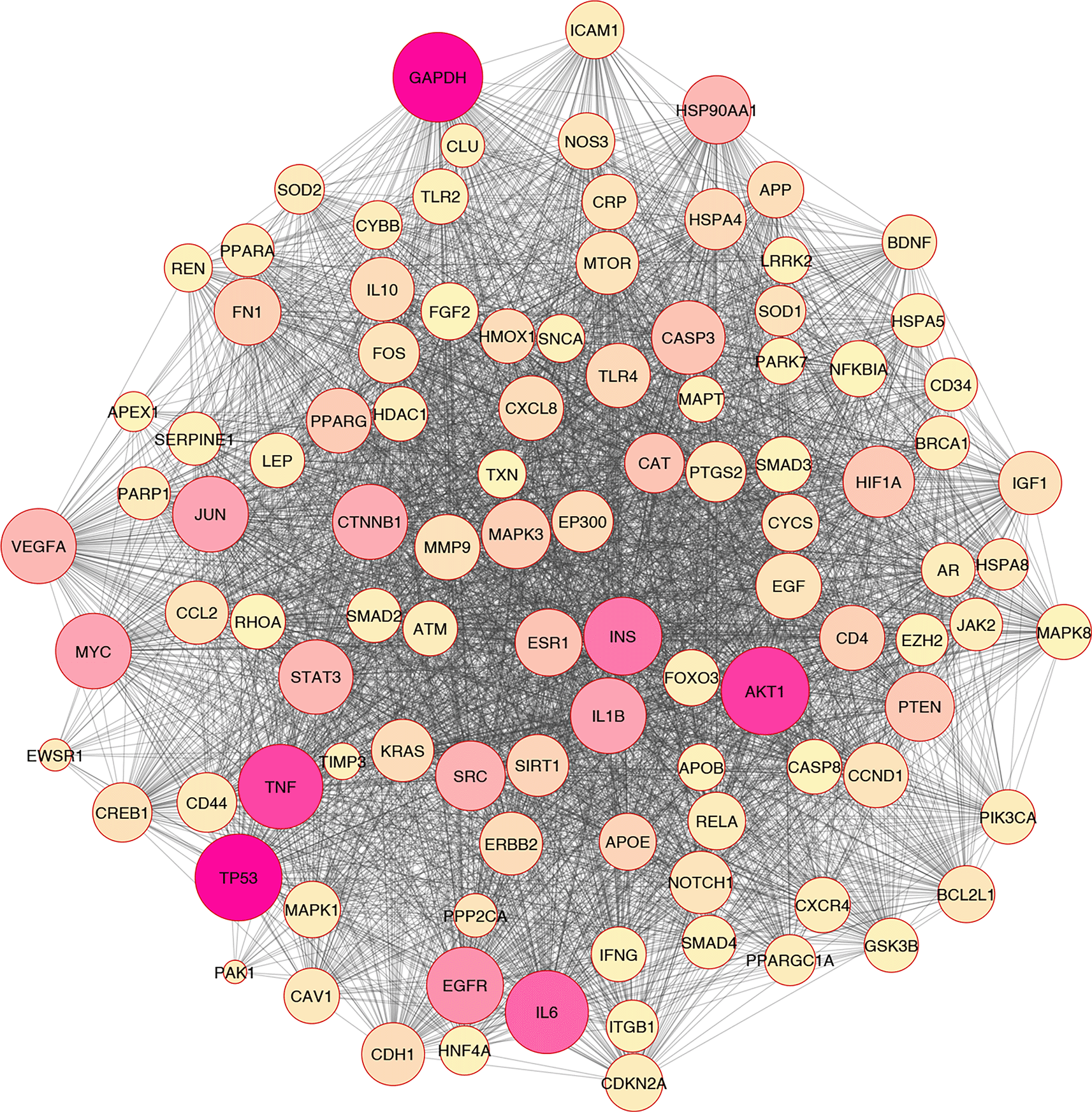
In silico and bioinformatics analysis have revealed that the hub genes involved in cell death, cytokine production, and oxidative stress pathways could be related to T2D (Figures 1–3). As a significant target in cardiac apoptosis, Cytochrome C (CyC), sirtuin 3 (Sirt3), B-cell lymphoma 2 (Bcl-2), and Fas cell surface death receptor (Fas) network may be essential for preventing heart failure.29 BCl-2 protein may inhibit the cell death apoptosis pathway and reduce cytochrome C activity in mitochondria. Moreover, apoptosis activators such as BAX (BCL2 Associated X) can increase cytochrome C secretion.50,51 The interaction between FAS and FAS ligands may mediate the apoptotic death of cardiac cells. Thereby, hyperglycemia prompts the increased ROS, releasing cytochrome C, mitochondrial malfunction, and damage to heart tissue in diabetic conditions.52–54
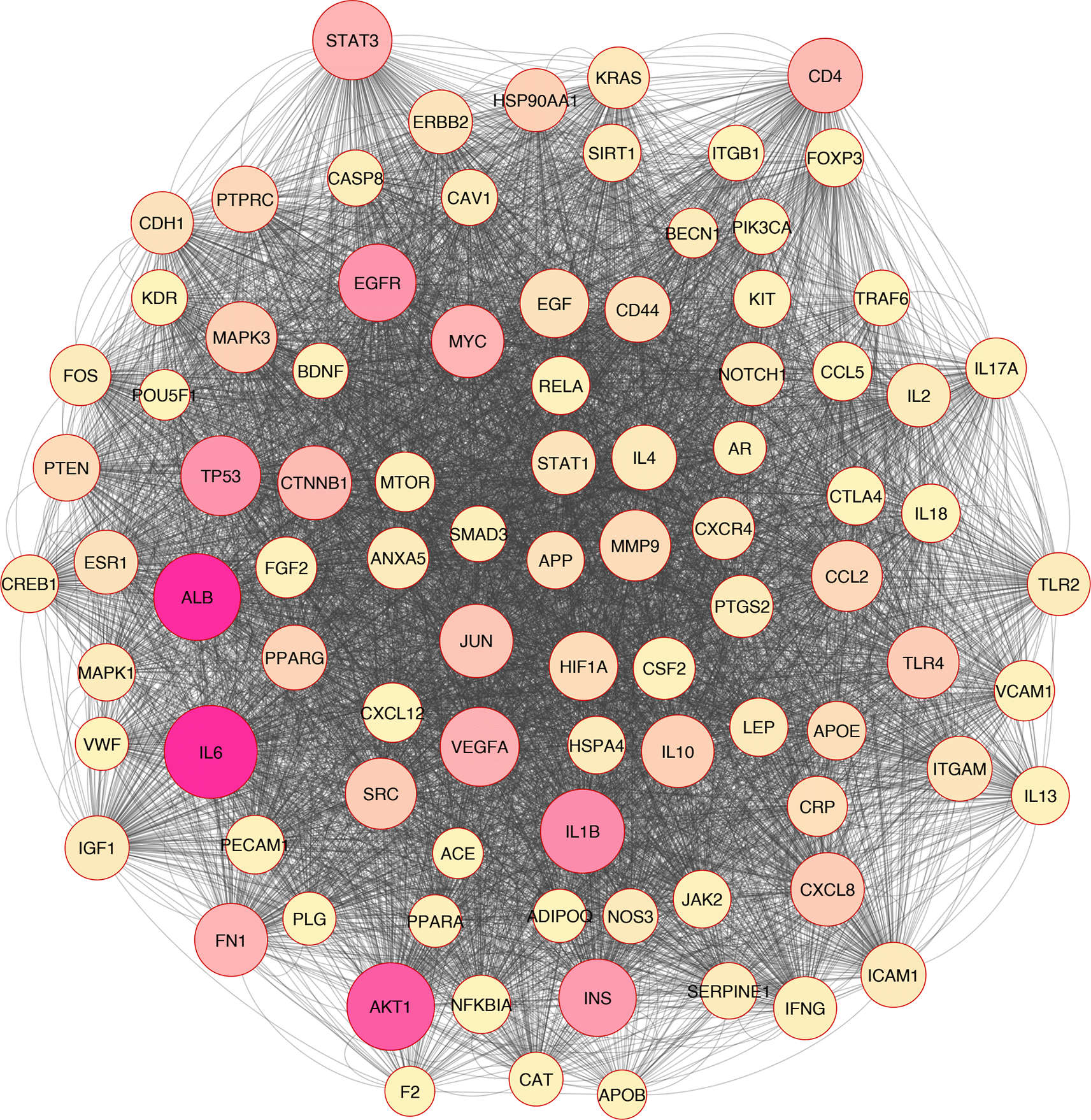
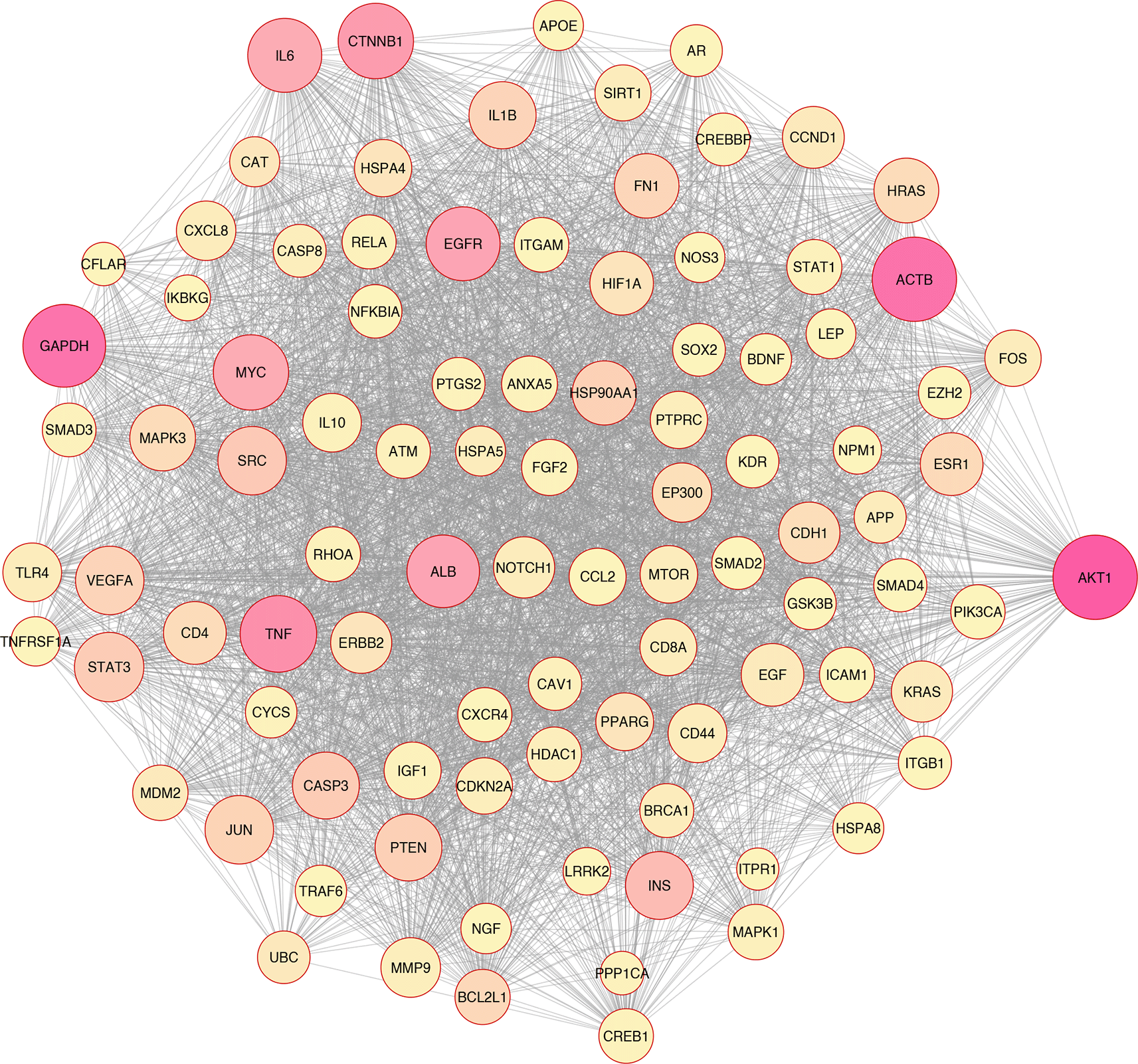
Based on the evidence, Sirt3 is identified as a coding gene that encodes class III histone deacetylases of the sirtuin family. Moreover, Sirt3 is exclusively found in the mitochondrial matrix membrane. Consequently, this gene may function as a master regulator under various circumstances and induce apoptosis.50,55
In addition, Sirt3 has been discovered as a gene with far-reaching effects on energy metabolism, aging, apoptosis, diabetes, cardiovascular diseases, and neurological illnesses.56,57 Enhancement of ROS production causes oxidative stress, which triggers several cellular alterations.43 Increased ROS generation and apoptotic activation in β-cells are hallmarks of diabetes, whether acute or chronic.58 Tissue damage in the retina, heart, nervous system, and kidneys may lead to apoptosis and necroptosis, all of which play critical roles in developing diabetes complications.59,60 Growing evidence has indicated that apoptosis, autophagy, necroptosis, and ferroptosis are the categories of cellular death61–67 (Figure 3).
It should be noted that cell death mechanisms have a diversity of functions. The nomenclature committee on cell death (NCCD) has spent developing criteria for cell death's morphological, biochemical, and functional classifications and descriptions.67 Apoptosis occurs following cytoplasmic shrinkage, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), apoptotic body formation, phagocytic activity, and degradation within lysosomes by neighboring cells.68
Type 1 cell death (apoptosis) occurs following cytoplasmic shrinkage, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), apoptotic bodies formation, phagocytic activity, and degradation within lysosomes by neighboring cells.48 Immense evidence has indicated that triggered Caspase is a crucial mechanism for inducing cell death.49,69 Multiple triggers operate on different cellular components in the intrinsic process of apoptosis, also called the mitochondrial pathway.50,70 As well as, after environmental damage, natural killer cells and macrophages release death molecules that, upon binding with death receptors (DRs) on the target extracellular membrane, trigger the extrinsic cascade of apoptosis by activating procaspase 8 to caspase 8. The DRs are a class of proteins that belong to the TNF (tumor necrosis factor) superfamily.51,71
On the other hand, saturated fatty acid lipid peroxidation is dramatically accelerated in the presence of iron, especially divalent iron. Cells generate ROS in addition to ATP during iron-dependent oxidative phosphorylation in the mitochondria.46 An oxidative stress response is triggered when the amounts of ROS in a cell are higher than its ability to neutralize them, and this can cause harm or death to the cell by directly or indirectly damaging significant molecular components, including proteins, nucleic acids, and lipids. Hence, increased levels of iron-dependent lipid peroxide cause ferroptosis cell death. Initiating ferroptosis requires reactive oxygen species production and iron availability.44,45
Moreover, according to recent investigations, autophagy has emerged as a critical position in preventing insulin resistance and death generated by oxidative stress in pancreatic islet beta cells.62 Antioxidant-rich proteins and organelles are degraded and recycled by autophagy to preserve cellular homeostasis. ER stress, starvation, and growth factor deficiency can all trigger this process.61,72,73 According to growing research, autophagic dysfunction has been linked to numerous disorders, including tumors, neurodegenerative diseases, and inflammatory diseases. Autophagy may have a role in the pathophysiology of metabolic diseases like diabetes by redistributing resources away from inefficient activities and toward crucial ones essential for life.
Necroptosis is the best-understood format of programmed necrosis death, exhibiting necrosis and apoptosis characteristics. TNF-1 (TNFR1) and Fas receptor ligands are involved in the cascade of necroptosis activation in response to high glucose stimulation.61 As well as necroptosis, damage-associated molecular patterns (DAMPs), nucleotide binding and oligomerization domain (NLRs), ripoptosome, and protein kinases R (PKR) complex are also involved in necroptosis activation. Apoptosis, necroptosis, or cell survival are all potential outcomes of TNFR1 binding to TNFs. Cell survival, apoptosis, or necroptosis are three distinct obligations that various signaling complexes can mediate following TNFR1 activation by TNFs binding. Based on the evidence, complex-I promotes survival, while complex-IIa promotes apoptosis.61 Necroptosis occurs as a result of the development of complex-IIb. Moreover, necroptosis is regulated by the receptor-interacting protein 3 (RIP3).61 In diabetes, RIP3 controls necroptosis by regulating RIP1 in a RIP-dependent way.74 RIP3 is triggered by self-phosphorylation to enhance MLKL phosphorylation after being recruited by RIP1 during high glucose exposure.66 These results in the cell membrane being permeable when MLKL oligomers translocate to the cell membrane, interacting with PIP2 lipids and cardiolipin. It has been discovered that CaMKII induces necroptosis. The myocardium contains a lot of CaMKII, which is usually inactive. Evidence has indicated that several factors, such as RIP1 phosphorylation, increased Ca2+ accumulation, and high glucose may activate necroptosis.74,75
These pathogenic alterations lead to necroptosis by activating the mitochondrial permeability transition (mPTP). Some evidence suggested that knocking down CypD, which increased the likelihood of mPTPs opening, could protect against RIP3-induced cardiomyocyte necrosis.76 Finally, the opening of the mPTP is brought about by the up-regulation of RIP3, which increases CypD phosphorylation and boosts PGAM5 expression. As the death signal was amplified, necroptosis occurred in the endothelial cells due to excessive mPTP opening.77
On the other hand, toll-like receptor 4 (TLR4) activation is another critical mechanism of ROS and hyperglycemia that can trigger apoptosis.78 A literature review revealed that the expression of the TLR4 is up-regulated in diabetic conditions, and TLR4 gene silencing may be able to regulate cardiac apoptosis in diabetes.58,79 Evidence has demonstrated an association between TLR4 and oxidative stress.80 As a result, the reduction of TLR4 leads to the dampening of stress responses, and caspase 3 in cardiomyocytes may be inhibited.81
Notably, the inflammatory mediators of diabetes, such as lipoprotein-associated phospholipase A2, adiponectin, tumor necrosis factor-α (TNF-α), high mobility group box-1 (HMGB-1), advanced glycation end products (AGEs), and chemokines, interleukin-1 (IL-1), interleukin-6 (IL-6), interleukin-8 (IL-8), and interleukin-18 (IL-18). Upregulation of these inflammatory mediators might activate nuclear factor κB (NF-κB), which induces cytokine storm and angiogenic mediators. Hence, accumulation of the ROS molecule and inflammation agents leads to cell death and impairs the function of the cell.58,82
Moreover, NO is an inflammatory and damaging component due to interactions with the intermediate factors of reactive oxygen species generally available in cells.83 NO/inflammation signaling cascade pathway may mediate several diseases, including lung squamous cell carcinoma, colon cancer, depression, and diabetes.84–87 One category of nitric oxide synthase factors, endothelial nitric oxide synthase (eNOS), plays a significant role in managing NO function by stimulating NO synthesis.88
Ischemia/reperfusion-induced acute kidney injury, ischemia/reperfusion-induced liver injury, hypertension, and steatohepatitis have been highlighted as associated disorders with the PPAR/eNOS axis.89–92 Even though the PPAR/eNOS axis mediates the inflammatory status involved in T2DM pathogenesis, the mechanism and vital molecules remain controversial.
Overall, based on the mentioned evidence, we could conclude that enhanced ROS molecules mediated the other pathomechanisms such as inflammation, apoptosis, autophagy, necroptosis, and ferroptosis. Furthermore, we found that oxidative stress correlated with inflammation and cell death based on the in silico analysis (Figure 2).
Interactions between genetic and epigenetic elements establish the pathogenetic state and may be affected by several regulatory elements.93 The length of non-coding RNAs is utilized for the recognition category of short non-coding RNAs and long non-coding RNAs (LncRNAs).73,74 MicroRNAs (miRNAs), one type of short non-coding RNA consisting of around 20–25 nucleotides, have a role in the biological processes by modifying the post-translational events of mRNAs.75 MiRNAs regulate gene expression by binding to specific seed sequences in 3′ untranslated region (3′UTR) of target genes. Moreover, miRNAs are considered significant biomolecules involved in the beginning, development, prognosis, diagnosis, and therapy of different pathophysiological indications due to their role as fine tuners of gene expression, control of signaling, and molecular-cellular processes.8,75 Typically a nucleotide sequence of over 200 bp displaying as long noncoding RNAs (lncRNAs) influences gene expression by many mechanisms.76 The linear and circular forms of lncRNAs are found in cells and the extracellular space.77 The RNAs that exist in a circular structure are referred to as circular RNAs (circRNAs). Furthermore, the annotated lncRNAs have been used to classify lncRNAs as the signal, decoy, guide, and scaffold molecules. Hence, non-coding RNAs are also interesting and attractive therapeutic targets since of their potential impact on various disease-related cellular processes.28
Growing evidence highlighted that lncRNAs have been recognized as significant transcripts involved in oxidative stress, inflammation, cell damage, apoptosis cell death, beta-cell function, insulin resistance/insulin secretion, metabolism, etc., and are now considered potential diagnostic biomarkers.78 Hence, long non-coding RNAs are potentially pivotal in oxidative stress, inflammation, and cell death studies. Only a significant portion of annotated lncRNAs have been shown to play critical roles in diseases associated with oxidative stress, such as those affecting the nervous, respiratory, metabolism, and cardiovascular systems.28 Based on the evidence, lncRNAs could target key hub genes related to the oxidation and antioxidation balance system, such as the ARE/Nrf2/Keap1 network.79 The most critical lncRNAs related to oxidative stress conditions could be pointed to the MALAT1, H19, SCAL1, NEAT1, GADD7, MACC1-AS1, ODRUL, LINC01619, LINC00963, FOXD3-AS1, and BDNF-AS.80 The previous review study briefly discussed lncRNAs Meg3, SNHG16, MALAT1, GAS5, HOTAIR, and CASC2's role in diabetic cardiovascular vascular damage through the oxidative stress response. Pei-Ming Chu et al. revealed that MEG3 targets miR-145/PDCD4 and HOTAIR by inhibiting miR-34a/SIRT1, miR-126/SIRT1, and PI3K/Akt could be regulated oxidative stress in diabetic cardiomyopathy complication status.28 Moreover, H19/miR-675 axis has been shown to modulate apoptosis cell death of cardiomyocytes by influencing voltage-dependent anion channel 1 (VDAC1), which is one of the several pathways involved in the progression of diabetic cardiomyopathy.81
Based on a systematic review and in-silico analysis, Cristine Dieter and colleagues revealed that lncRNA ANRIL, HOTAIR, MALAT1, MIAT, and KCNQ1OT1 are up-regulated in diabetes Mellitus patients compared with control subjects, and MEG3 is down-regulated, consistently.77 Based on these results, we concluded that lncRNAs can mediate and manage the oxidative stress signaling pathway in type 2 diabetes.
Epidemiological evidence has indicated that environmental risk factors such as alcohol, smoking, high-fat diet, and physical inactivity could contribute to oxidative stress. In addition, environmental factors may play a crucial role in the pathogenesis of heart failure and non-communicable diseases.83 Some studies have demonstrated that ROS might regulate the activity and mechanism of tissues, including muscle, liver, bone, intestinal, and brain.83–85 Most tissues, especially muscle and adipose tissue, continuously produce ROS at low-level exercise intensity.85 ROS production indirectly or directly influences muscle activity by several mechanisms (metabolism, calcium homeostasis, contractility, and excitability), which causes skeletal muscle exhaustion during intense exercise.19
Moreover, long exercise training, overtraining syndrome, and exhausting exercises increase ROS production.86 In contrast, prolonged and low/moderate-intensity training ameliorates endogenous antioxidant status. Based on the evidence, physical activity could up-regulate mitogen-activated protein kinase (MAPK), and NF-κB induces several hub genes and proteins involved in the homeostasis, inflammation, and antioxidant/oxidative intracellular.87
Physical activity and dietary modifications are two of the most effective nonpharmacological strategies for many chronic conditions, especially cardiovascular conditions.85 In addition, ample evidence has revealed that exercise training and physical activity positively affect the cardiovascular system by improving oxidative stress, apoptosis, mitochondrial biogenesis, metabolism, and autophagy.88 Endurance training could up-regulate the expression level of the PPARγ/Pgc-1α/Fndc5 pathway in the heart and muscle mice. Evidence indicated that FNDC5 (fibronectin type III domain containing 5) and its cleavage type, irisin, might have an essential role to play in energy regulation and metabolism. Several studies demonstrated that PPARγ/Pgc-1α/Fndc5 could trigger by physical activity and exercise training, which induced the white adipose tissue's browning, and enhanced weight and energy expenditure.89
Moreover, Abedpoor and colleagues revealed that consuming the branched-chain amino acids (BCAAs) and endurance training regulated the PPARγ/Pgc-1α/Fndc5 expression. Based on this study, BCAAs and endurance training improved the mitochondrial biogenesis genes such as Tfam, Cox4i1, ATP5a1, ATP5b, and Sirt1.89 Interestingly, Bahadorani et al. demonstrated that BCAAs and physical activity modified the percentage of sperm lipid peroxidation and sperm parameters.90 On the other hand, ample evidence indicated that a High-fat diet and diet enriched in advanced glycation end products (AGEs) could dysregulate the detoxification status and increase the oxidative stress in the cells.84,91 Besides, endurance exercise and leucine consumption modified the hub genes and lncRNAs involved in the brain-gut axis in mice with depression-like behaviors.76 The authors found that the Kdr/Vegfα/Pten/Bdnf expression level was regulated by endurance exercise and leucine consumption in the hippocampus region and ileum tissue.
In addition, several evidence indicated that lifestyles such as physical activity and unhealthy diets could impact fertility and sperm function.92,93 High-fat diet and diet enriched advanced glycation end products dysregulated the function, parameters, and intracellular reactive oxygen species of sperm.92 Furthermore, high-fat diet-enriched AGEs enhanced the inflammation and apoptotic signaling pathway in the intestines of the mice.91 Based on these data, consuming the high-fat diet enriched significantly increased the IL-6, AGER (advanced glycation end products receptor), ZO-1, P53, and NF-κB. In this study, the relative expression of the Sirt-1 and Ddost were decreased by consumption of the high-fat diet enriched. In the other study, a high-fat diet enriched with AGEs significantly increased the relative expression of the IL-1ß, which induced inflammatory bowel disease (IBD).91 Based on these data, we could conclude that physical inactivity and unhealthy food enhanced the inflammation and oxidative stress in the whole body.
Epidemiology studies revealed that a sedentary lifestyle and ultra-processed foods such as AGEs-rich meals and high carbohydrates distributed the defense mechanisms against oxidative stress, including the Nrf2-Keap1 signaling pathway and downstream genes such as HO1, Nqo1, Txn, and Gpx1. These hub genes could regulate antioxidant response elements. AGEs rich meals and physical activity significantly dysregulated the expression level of the Nrf2 HO1, Nqo1, Txn, Gpx1, and antioxidant capacity of the liver in diabetic mice. Notably, physical activity and herbal medicine reversed these hub genes and improved the antioxidant capacity, insulin and glucose concentration, and liver damage in diabetic mice. Impaired muscle mitochondrial function and biogenesis could be the other side effect of the sedentary lifestyle and ultra-processed foods. Hence, the mitochondrial markers were significantly decreased in response to a high-fat diet and ultra-processed foods. These results show that the relative expressions of the Cpt2, Tfam, Pgc-1α, mt-Nd1, mt-Nd5, Ndufa2, Cox8b, Cox5a, mt-Co2, and mt-Co1 were dysregulated. Notably, exercise training and dietary phytochemicals modified and improved the relative expression of genes related to the muscle mitochondrial markers (function and biogenesis) and β-oxidation.
Overall, based on these studies, we could conclude that environmental and exogenous factors have a substantial role in regulating oxidative stress of the cell. Moreover, environmental risk factors could raise ROS production due to oxidative stress, which induces several signaling pathways, including apoptosis, inflammation, autophagy, necroptosis, and ferroptosis.
Fatemeh Hajibabaie and Navid Abedpoor conducted the study design. The data mining was evaluated by Fatemeh Hajibabaie, Navid Abedpoor, and Faranak Aali. The manuscript was written by Fatemeh Hajibabaie, Navid Abedpoor, and Faranak Aali and was approved by Navid Abedpoor. The authors declare that all data were generated in-house and that no paper mill was used.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)