Keywords
obesity, prevention, short-chain fatty acids, propionate, gut microbiota
This article is included in the All trials matter collection.
obesity, prevention, short-chain fatty acids, propionate, gut microbiota
Overweight and obesity affect over 68.2% of men, and 60.4% of women in the UK and drive the prevalence of several common co-morbidities, including type 2 diabetes, cardiovascular disease and cancer. Although significant advances have been achieved in the management of obesity, there has not been the same level of research activity in obesity prevention. Once an individual becomes obese the probability of returning to normal body weight is extremely low (1 in 210 for men and 1 in 124 for women).1
Weight gain occurs commonly throughout adulthood and younger adults are at the greatest risk of substantial gains in body weight, according to a National Health and Examination Survey (NHANES) of adults aged 25–74. Major weight gain over 10 years is categorised as a gain in Body Mass Index (BMI) ≥5 kg/m2, this weight gain was highest in those aged 25–35 years.2 Whilst a relatively modest weight gain of 1 kg over a single year would present a very low risk to health in young adults, the accumulated weight gain over a decade or longer leads to a clear deterioration of cardiovascular and diabetes risk factors. For example, the 10-year Coronary Artery Risk Development in Young Adults (CARDIA) study demonstrated that weight gain during early adulthood produced measurable adverse changes in blood lipids, fasting insulin, and blood pressure, irrespective of race or gender.3
Epidemiological and experimental studies have demonstrated an inverse association between dietary fibre intake and body weight gain.4,5 Current fibre intake is approximately 17–20g/d,6 well below the 30g recommendation.7 The daily intake of dietary fibre in the UK has not increased for 10 years.6 Although the mechanisms surrounding how elevated dietary fibre intake affects energy balance are not fully understood there is evidence that the fermentation of dietary fibre in the colon by the microbiota produces short chain fatty acids (SCFA) that stimulate the release of anorectic gastrointestinal hormones PYY and GLP-1.8,9 In previous research, a methodology for delivering the SCFA propionate to the colon was developed by esterifying it to a non-digestible fructooligosaccharide. It has been demonstrated that this novel food ingredient, inulin propionate ester (IPE), stimulated the release of PYY and GLP-1 and lowered energy intake.10 The proof of principle study demonstrated that over six months, the consumption of 10 g of IPE daily significantly prevented weight gain.10
The current study aims to investigate the impact of IPE on the prevention of weight gain in young adults aged 20 to 40 years over a period of 12 months.
This clinical trial is a randomised, placebo-controlled, double-blind trial to investigate the efficacy and safety of inulin propionate ester (IPE) versus the inulin (fermentable oligosaccharide) control upon weight gain prevention. Participants will be randomised to either IPE or inulin for 12 months. This trial will be performed at two UK sites: Imperial Clinical Research Facility (CRF) in London - Imperial College Healthcare NHS Trust and Glasgow CRF - NHS Research Scotland. This trial was registered with International Standard Randomised Controlled Trial Number (ISRCTN) registry (ISRCTN16299902) on 1st March 2018 (https://doi.org/10.1186/ISRCTN16299902), before recruitment commenced. Participants will be invited to attend a screening, baseline/randomisation and all subsequent trial visits at two, six and 12 months will be conducted at the CRF of each participating site. See participant timeline, Figure 1. Participants recruited at Imperial will also be invited to participate in a mechanistic sub-study and if they agree these assessments will take place at baseline and 12 months at the Imperial CRF.
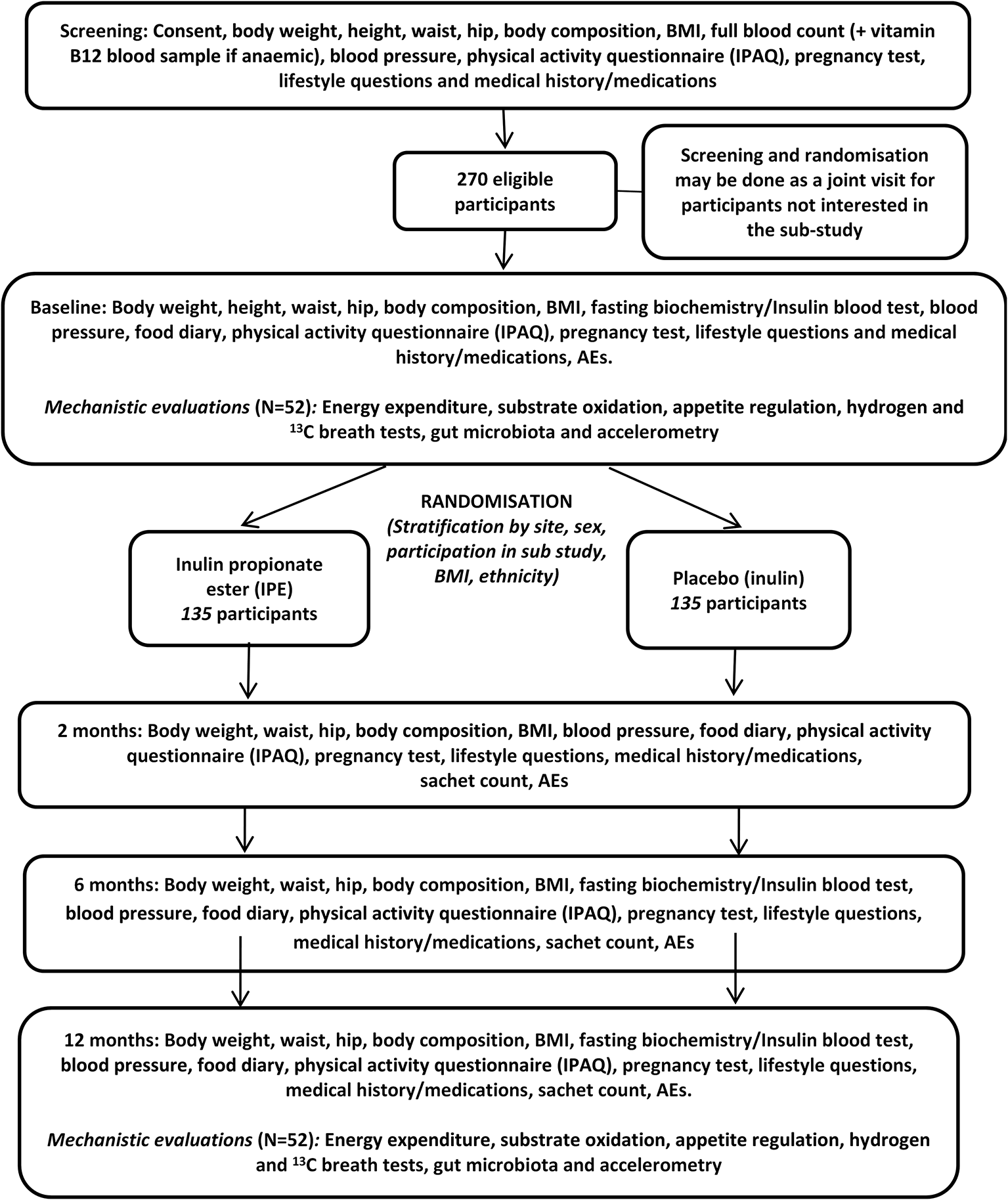
Inclusion criteria
• Males and females aged 20–40 years
• Body Mass Index (BMI) of 24.0–27.0 kg/m2 if of South-Asian ethnicity or 25.0–30.0 kg/m2 if non-South-Asian
• At least one of the following:
• On same medication for >3 months at point of screening
• Written informed consent
Exclusion criteria
• Diagnosed chronic disease Type 1 and 2 diabetes, cancer, renal failure, heart disease, organic acidaemia (propionic acidaemia, methylmalonic acidaemia)
• Diagnosed gastrointestinal condition (coeliac disease, inflammatory bowel disease and irritable bowel syndrome)
• Previous bowel reconstruction surgery
• Pregnancy or lactation
• Use of antibiotics in the past three months
• Vitamin B12 deficiency (<160 ng/L)
• Taking part in a weight loss program or consuming a weight loss product
• Have lost 3 kg or more in the last three months
• Gastrointestinal upset in the last two weeks
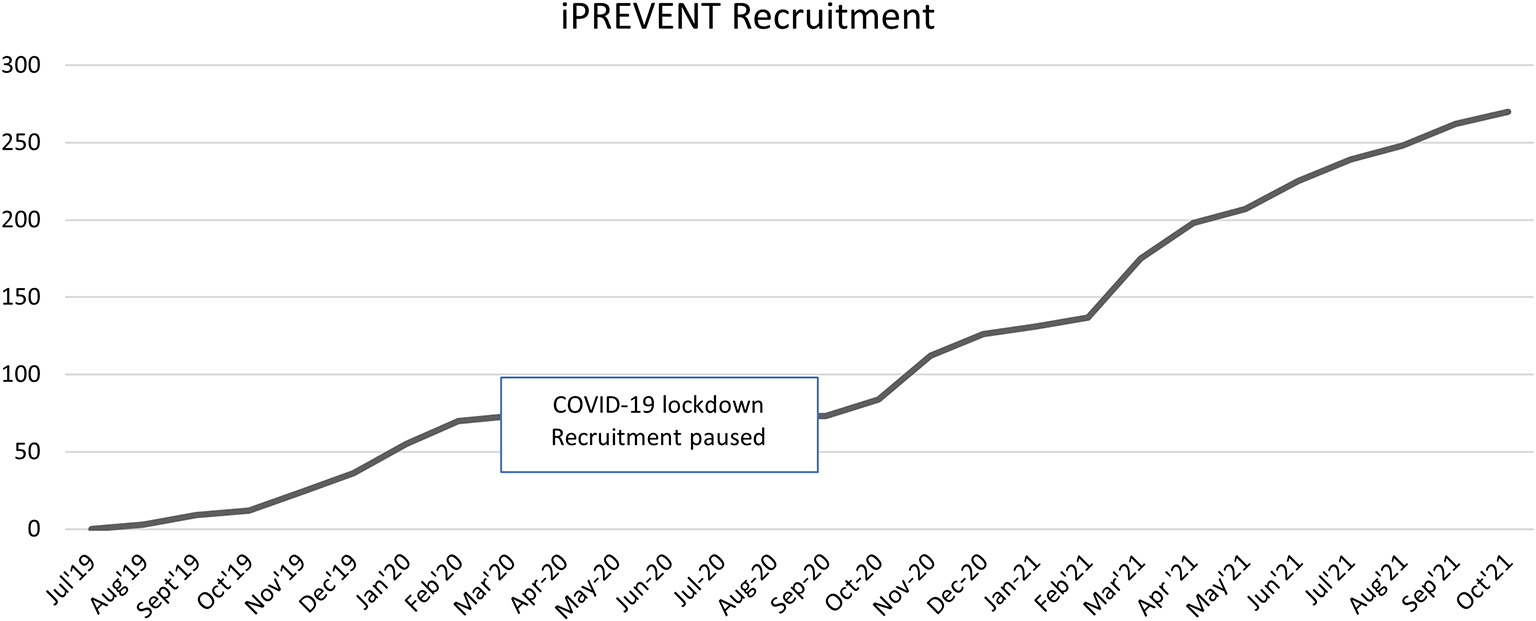
A wide variety of recruitment methods were used for this clinical trial, including recruitment via GP practices and within NHS trusts, newspaper adverts, pop-up events and posters, amongst other methods. Recruitment was completed in October 2021 (Figure 2).
Randomisation will be undertaken using minimisation with a random element to balance the arms by research centre, sex, and BMI within ethnicity (South Asians: 24.00–25.49 kg/m2 and 25.50–27.00 kg/m2/non-South Asians: 25.00–27.49 kg/m2 and 27.50–30.00 kg/m2) and whether they take part in the mechanistic sub-study. Minimisation will be conducted by researchers using sealed envelope software (Open-source software, www.sealedenvelope.com).
Participants will receive blinded and identical-looking trial intervention of either IPE or inulin control. Both IPE and inulin present as white powders and will be delivered in pre-packed plain foil-backed sachets. Participants will be identified with a unique trial identifier and each IPE or control sachet will be identified with a unique treatment code linked to the allocation and trial identification (ID). The treatment code will not be broken except in medical emergencies or if expedited reporting to the Research Ethics Committee (REC) of an unexpected and related Serious Adverse Event (SAE) is required.
At each trial visit, trial researchers will conduct all necessary measurements and sample collections as described in Tables 1 and 2. In the main study, written informed consent is given at the screening visit, blood samples are taken (excluding two-month visits), blood pressure, body weight, body composition and compliance are measured and occurrence of Adverse Events (AEs) and SAEs are documented at all study visits. In the sub-study breath, stool, urine and blood samples are taken and breath hydrogen, energy expenditure, subjective appetite and energy intake are measured.
Primary outcome
The primary outcome is weight gain from baseline to 12 months.
Secondary outcomes
• Occurrence of AEs and SAEs
• Changes in fasting biochemistry:
• Changes in blood pressure
• Changes in body weight
• Changes in waist/hip/BMI/body composition measurements
• Changes in compliance
Mechanistic outcomes
• Gut microbiota: 16S rRNA profiles from stool samples
• Impact on neuroendocrine cell number: Proliferation in intestinal organoids using the level of SCFA and other metabolites identified from nuclear magnetic resonance spectroscopic analyses of stool
• Appetite regulation: Measured by visual analogue scales (VAS), food diaries, ad libitum intake, and appetite-regulating gut hormones PYY, GLP-1, Gastrin and CCK
• Energy expenditure: Open-loop indirect calorimetry
• Hepatic lipid metabolism: Stable isotope tracers of fat oxidation (13C palmitate) and De Novo Lipogenesis (DNL)
• Total body water through dilution analysis of 2H2O as already applied.
Proposed amendments to the protocol and aforementioned documents will be submitted to the REC for approval as instructed by the Sponsor. Amendments requiring REC approval may be implemented only after a copy of the REC’s approval letter has been obtained. The regulatory authorities and REC will be sent annual progress reports and informed about the end of the trial, within the required timelines. Table 3 details the amendments that have been submitted/approved by the REC so far.
The following amendments have been submitted/approved by the REC so far.
Participants may discontinue trial intervention for the following reasons:
• At the request of the participant
• Due to an Adverse Event/Serious Adverse Event
• If the investigator considers that a participant's health will be compromised due to AEs or concomitant illnesses that develop after entering the trial.
If a participant permanently discontinues the trial intervention, they will be invited to continue to attend trial visits, if possible, to allow for the collection of key outcome and safety data.
Discontinuation of trial intervention and procedures can occur for the following reasons:
If a participant withdraws from trial procedures, an attempt will be made to obtain self-reported body weight (primary endpoint) at the point of withdrawal and at the final study visit time point. If the participant does not agree for data and samples collected to be retained, the samples must be destroyed and data excluded from the analyses.
If the participant withdraws consent to further be contacted at all for the study purposes, no attempts to obtain a self-reported body weight for the primary endpoint will be made.
All serious and non-serious AEs will be reported on the trial database, from the point of screening until the end of the study, except for elective medical procedures. All SAEs will be reviewed by both the local investigator and chief investigator or a designated medically qualified representative to confirm ‘expectedness’ and ‘causality’. SAEs are assessed on whether they are possibly, probably or definitely related to the trial protocol/intervention. An unexpected event would be a type of event that is not listed in the trial protocol or document associated with the intervention, as an expected occurrence. Expected AEs for this trial are gastrointestinal effects. If the investigator becomes aware of safety information that appears to be related to the trial, this should be reported to the trial coordination centre and sponsor.
PPI panels were held during the design and management and recruitment phases of this clinical trial.
Recruitment for this study was completed in October, the study is currently in follow-up phase and 12-month visits will be completed by the end of October 2022.
Sample size calculations
In the randomised proof of concept trial, the difference between arms in the change in body weight over 24 weeks was 1.4 kg (95% CI: -0.3 to 3.1), p = 0.099. Using a Bayesian method recommended for preliminary trials in which evidence in the 95% CI is translated into probabilities,11 there was a 95% posterior probability of an underlying positive between-arm difference favouring the intervention. The posterior probability of intervention-favouring differences greater than 1 kg, 1.5 kg, and 2 kg were respectively 69%, 47% and 25% based on 24-week intervention. The difference increased in magnitude through successive eight-week, 16-week, and 24-week time points. By 24 weeks there were significant reductions in the proportion of intervention participants gaining 3%, and 5% of body weight from a mean baseline of 90 kg. A 2 kg between-arm 12-month effect size was therefore chosen. This agreed with a weight gain prevention trial over nine months in young adults12 which aimed to detect a 2 kg effect and achieved 4.3 kg, with a pooled standard deviation (SD) for body weight change of 4.35 kg, and 81% retention.
On this basis a sample size of 270 randomised participants (135 per arm) was chosen to provide 90% power to detect a 2 kg difference between arms in mean body weight change over 12 months using a two-sided 5% level significance test, assuming a 4.35 kg SD and with 25% dropout allowance (68 participants). The sample size was calculated using R Project for Statistical Computing (RRID: SCR_001905).
For the mechanistic study, 34 volunteers (17 per group) would provide sufficient statistical power to detect a 15 pmol/L effect size in PYY and GLP-1 concentrations between groups, with 90% power, 5% significance level, SD 13 pmol/L. These differences are based on previously published findings that report enhanced gut hormone release following IPE supplementation.9,13 A subsample of 52 volunteers (26 per group), was chosen, to allow a 70% retention rate.
Data analysis plan
This trial has both a pragmatic element, to answer the question of whether the policy of prescribing and uptake of inulin propionate ester as specified in the trial will reduce further weight gain compared with control, and an explanatory element to understand the mechanisms of the causal pathway of such body weight change and any limitations from compliance. Therefore, analyses will be primarily on an ‘Intention to Treat’ basis. Secondarily, analyses will incorporate mechanistic sub-study data, and use this to understand the ‘Intention to Treat’ effect estimated in the trial.
Statistical methods for the analysis of the study endpoints are described below. IBM SPSS Statistics (RRID: SCR_019096), version 28.0.1, will be used for all statistical analyses. A separate detailed Statistical Analysis Plan (SAP) has been prepared and approved by the Trial Steering Committee.14 This contains the rationale for the methods chosen and the assessment of their assumptions. It includes pre-specifying the handling of covariates and missing data, compliance analyses, and the primary estimand. Changes to the plan can be revised and re-approved by trial oversight committees prior to database lock.
Any deviations from the SAP during analysis after the data lock will be documented and signed off by the statisticians and CI and filed in the statistics section of the Trial Master File (TMF) which will be merged with the main TMF at the end of the study. The final approved SAP, being in accordance with the protocol, takes precedence for undertaking the analysis of the main trial paper.
Primary endpoint analysis
The analysis of the primary endpoint will incorporate the earlier correlated interim measurements of body weight in a linear mixed effects model and will adjust for baseline continuous body weight and other categorical randomisation stratifiers with further specification of the role of timepoint, and correlation structure, detailed in the SAP. The implicit ‘missing at random’ assumption will be challenged through a set of sensitivity analyses.15 As these involve all randomised participants, this is therefore an Intention to Treat Strategy.16
Secondary endpoints analysis
Where possible, continuous secondary endpoints will be adjusted for their baseline to improve the precision of estimated intervention effects. Repeated measures will be analysed using linear mixed-effects models adjusting also for randomisation stratifiers. Comparisons between arms for binary outcomes will be summarised as differences in proportions. Confidence intervals of 95% will be used to make inferences from estimated effect sizes.
Ethical and safety considerations
The investigator(s) will ensure that this trial is conducted in full conformity with the 7th revision of the 1964 Declaration of Helsinki. The trial will be conducted following the guidelines laid down by the International Conference on Harmonisation for Good Clinical Practice (ICH GCP E6 guidelines and Addendum ICH GCP E6 (R2)). The trial protocol and participant-facing documents have been reviewed and approved by the London Hampstead REC Reference 14/LO/2004). Clinical trial authorisation from the Medicines and Healthcare products Regulatory Agency (MHRA) is not required as the study intervention is a dietary supplement.
Data management
All personal identifiable data, including screened patients, will be kept securely in the local site files and will not be uploaded to the main trial database. The InForm™ trial database will be used for all visits data entries. All data recorded in the eCRFs will be signed off by the site investigator. The central coordinating site will visit local recruiting sites to ensure compliance with the protocol, good clinical practice and local regulatory compliance.
Dissemination policy
All publications and presentations relating to the study will be authorised by the Trial Management Group. Authorship will be determined according to the internationally agreed criteria for authorship (www.icmje.org). Authorship of parallel studies initiated outside of the Trial Management Group will be according to the individuals involved in the project but must acknowledge the contribution of the Trial Management Group and the Study Coordination Centre.
Trial organisation and committees
A fully independent Data Monitoring and Ethics Committee (DMEC) has been set up to monitor progress, participant safety and any ethical issues involved in this trial. They review trial progress, recruitment rates and safety data. Meetings are approximately six-monthly. Furthermore, a Study Advisory Group consisting of public representatives was also set up. The committees were set up and coordinated in accordance with the funder guidelines. Upon completion, the trial results will be published in peer-reviewed journals and presented at national and international scientific meetings. A Trial Steering Committee (TSC) has been convened to provide overall supervision of trial conduct and progress. The TSC meet approximately six-monthly throughout the trial. DMEC reports include details of recruitment, randomisation balance and stratification effectiveness, baseline characteristics, unblinding, withdrawals, compliance, concomitant medications, efficacy, mediators, and Aes.
To date, few randomised controlled trials focus on the prevention of obesity, especially in those most at risk of weight gain.2 High intakes of dietary fibre are associated with lower body weight.13 Evidence suggests that this is due to the generation of SCFA from colonic fermentation of dietary fibre by the gut microbiota.8 Furthermore, there is prior evidence to suggest that IPE could have a substantial impact on lowering individuals’ weight gain trajectory leading to a lowering of their long-term risk of obesity-related co-morbidities.9,12 This trial will explore all aspects of the novel food ingredient IPE; assessing efficacy via the primary endpoint of weight change, whilst attempting to identify potential mechanisms via the sub-study and accounting for compliance, tolerability and safety. The latter is an aspect that often goes unreported in similar trials; however, it is required to build a multifaceted evaluation of any intervention. This study methodology is not without limitations, primarily, narrowing the age range of the study sample may mean that results cannot be translated to the wider population. Additionally, the aforementioned scarcity of studies exploring the effects of SCFA on human populations meant that researchers had few studies to refer to when calculating population sample size, which may lead to a difference in predicted versus actual study power. However, it should be acknowledged that the study duration and sample size are greater than similar previously conducted studies.9,17–20 This randomised controlled trial aims to build upon previous literature to support evidence for the delivery of propionate to the colon for the prevention of weight gain.21
Obesity research has limited emphasis on prevention. Therefore, this randomised controlled double-blinded trial was designed to investigate whether inulin-propionate ester prevents further weight gain in adults most at risk of substantial increases in their BMI. Furthermore, the study adopts novel approaches to determine the underlying mechanisms influencing these beneficial effects.
Mendeley Data: Extended data for ‘Increase in colonic Propionate as a method of prEVENTing weight gain in adults aged 20-40 years (iPREVENT): A multi-centre, double-blind, randomised, parallel-group study to investigate the efficacy of inulin-propionate ester versus inulin (control) in the prevention of weight gain over 12 months’ statistical analysis plan’.
• Data file 1: iPREVENT Statistical Analysis Plan. pdf. https://doi.org/10.17632/n33kky5dww.214
• Data file 2: iPREVENT Protocol v5.0 15Feb2021.pdf. https://doi.org/10.17632/5n7h4xfz2n.1.21
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0)
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)