Keywords
Chimerism, de novo assembly, differential expression, read mapping, contig annotation, Drosophila melanogaster
This article is included in the Cell & Molecular Biology gateway.
This article is included in the RPackage gateway.
Chimerism, de novo assembly, differential expression, read mapping, contig annotation, Drosophila melanogaster
In the absence of a closely related reference set of transcripts that describes what may be expressed within a transcriptome, de novo assembly can be a pivotal point for transcriptomic experiments utilizing short-read RNA sequencing (RNA-Seq) data.1–3 The general goal is to increase understanding of how cells, tissues and organs develop,4,5 adapt,6,7 function8,9 and interact10,11 within their respective environments under varying conditions. This can be achieved through the characterization of expressed genes,1,12 the identification of differentially expressed genes13 and genomic level annotation using expressed transcripts as a guide,14–16 along with other types of RNA-Seq data analysis.17 In experiments involving short-read RNA-seq data, de novo assembly refers to the construction of a set of contigs from the short-read data that can be used as a reference set, often for the characterization of transcript expression profiles.18,19 Transcriptomics experiments have an impact across the entire living world, including host-pathogen interactions,20,21 the development of diseases such as cancers,22 diabetes,23 heart disease24 and Alzheimer’s,25 diseases associated with ageing,26 as well as animal27,28 and plant29,30 domestication; the latter requiring persistent alterations selected for through many generations. From an early stage, much effort has been invested in the optimization of experimental design and data analysis strategies.31 For example, a source of error that arises involves base calling during the sequencing process,32,33 and there are approaches available for read trimming34 and error correction35–37 that combine to form effective solutions.
A less obvious and less explored source of error is the erroneous chimeric sequences that can be introduced during the de novo assembly process.38–42 These are distinct from those introduced during library preparation.38,39 Non-chimeric contigs are assembled sequences that accurately represent expressed transcripts. Erroneous chimeric contigs occur when two or more fragments of DNA are incorrectly joined together. Primarily, the possibility of chimeric contigs arises when assembling portions of the read data representing increased biological complexity,40 such as transcripts expressed from multi-gene families.41,42 Many of the fundamental approaches to short-read assembly, including graph, reference and the overlap based methods, were developed in the early days of RNA-Seq read data analysis.43,44 The range of approaches developed, as well as the various parameter options explored,45–47 indicate the importance of this process. Combined with the variable results achieved,48–50 it is evident that a consensus “best approach” has not been resolved. More recent developments that incorporate the usage of long reads,51–53 in conjunction with long-read error correction54–56 and isoform characterization,57–59 will inevitably minimize the problem of chimeras in future studies. However, in the meantime, there are still many short-read datasets being generated, and there is a vast repertoire of archived short-read data that has been nearly two decades in the making.60 Thus, short-read RNA-Seq data yet possess scientific potential,61,62 and strategies improving its analysis are still relevant.
In relation to graph-based approaches, such as those implementing de Bruijn based strategies, including Trinity,63 rnaSPAdes64 and ABySS,65 as well as those creating networks based on splice sites identified following read mapping, such as TransComb,66 Cufflinks67 and StringTie,68 the ability to construct sets of contigs that represent the majority of transcripts present has been demonstrated.48–50,69,70 Potential has also been shown for graph-based approaches to provide detailed information on chimerism derived directly from the assembly process.40 During assembly, many graphs are constructed, and each aims to represent transcripts derived from single gene family.71 For graphs representing complex families, path choice during contig construction increases. This leads to the possibility of chimeras being produced in accordance with one of three broad categories: (i) over extension, (ii) increased sequence variation within regions and (iii) erroneously swapped regions.40,72 When reads are mapped to reference sets containing such chimeras, per-transcript counts values are affected.19,73 This increases the level of ambiguity within transcript expression profiles that are dependent on such counts.74,75 Additionally, it is these read count values that tools such as DESeq276 and EdgeR77 rely upon in order to characterize differential expression patterns. Given that short-read RNA-Seq platforms and de novo assembly software have been maturing for well over a decade,78,79 it is unfortunate that chimerism within assembled contigs, as well as the associated effects on read mapping and transcript expression profiling, have been under-characterized. A contributing factor is likely to be the difficulty in distinguishing between the extensive biological noise present within the transcriptome80,81 and artificial noise, including that of the erroneous chimerias created during assembly.72,82–84
Here the effects that chimerism has on read mapping and differential expression analysis are explored using both simulated and real data. To aid this a tool called ChimSim, which takes a base set of reference transcripts and introduces a user-specified proportion of chimerism, was developed. Using ChimSim reads simulated off a pre-defined base set of reference transcripts, for example - one of the many species-specific cDNA libraries available from Ensembl,85 can be mapped to corresponding modified sets containing incrementing portions of chimerism, following which mapping success relative to each transcript present was measured. In this study transcripts present within the cDNA library of Drosophila melanogaster (fruit fly) were used as the base set. When reads are simulated to allow for multiple replicates divided into two conditions, one of which reflects a pattern of read over representation across one thousand randomly selected transcripts, the effects of chimerism on the identification of over-expressed transcripts can be explored. To do this, a differential expression experiment was performed iteratively, where within each iteration, the extent of chimerism present within the modified reference set used for mapping replicates within each condition, prior to analysis with DESeq2,76 was incremented. Finally, we generated an expectation of the relationship between modified base sets containing incrementing portions of chimerism and the underlying fruit fly base set from which the modified sets were created. At each increment, this relationship was summarized by r-squared values describing the correlation between alignment and base set transcript lengths relative to matches identified, using megablast,86 between the base set and modified set. The same metric was then obtained for contig sets assembled using three graph-based assemblers CStone,40 Trinity63 and rnaSPAdes,64 in relation to the underlying fruit fly cDNA base set from with the input reads were simulated. By comparison back to the background distribution created, involving explicitly defined levels of chimerism, an estimate of the extent of chimerism present within assembled contig sets across multiple replicates could be inferred. In relation to the latter, assemblies were also created, and r-squared values calculated for contigs produced from RNA-Seq reads sequenced from fruit fly, obtained from a study on alternative splicing.87 These too were compared to the background distribution describing the correlation between alignment lengths and base set transcript lengths at incrementing levels of chimerism.
ChimSim is a tool that takes a set of sequences as input and creates an output set where a user-specified portion of the sequences is altered to be chimeric. ChimSim is written in Java and runs on operating systems with installed Java Runtime Environment 8.0 or higher (GNU General Public License v3.0). An executable version of ChimSim, as well as complete parameter descriptions, test data and source code, are available at: https://sourceforge.net/projects/chimsim/. An input reference set, for example, could consist of a set of expressed transcripts such as those available for varying species on Ensembl,85 or a compiled set of sequences manually defined by the user. Chimerism is introduced in accordance with one of the following three broad categories: (i) Over extension - transcripts have a region selected from a randomly chosen input transcript appended to them. The extent of over extension is selected at random from a range of between a minimum value (default: 100) and the length of the randomly selected transcript. (ii) Windowed Variation - windows are placed evenly along selected transcripts within which random variation is introduced. Window length (default: 200 nt), the number of windows (default: random between 1 and 5) and the level of divergence (default: 0.1, i,e, 10% of sites in window) can be defined by the user. If the length of a transcript is shorter than that of the combined length of the windows, then overlap is permitted. (iii) Window Shuffling - windows are created a similar manner to that described for (ii), but instead of variation being introduced, for each window a fragment of length equal to that of the window is selected at random from a different transcript and used to replace the region defined by a window. ChimSim outputs a text file containing the titles of transcripts selected to become chimeric, as well as the type of chimerism introduced.
The basic command to generate a set of modified transcripts from an input base set, where the default 10% of transcripts within the output will be chimeric and in accordance with default parameters, is: java -jar ChimSim.jar -ref_set path-to-ref-set.fasta.gz -gz true -out_dir path-to-out-dir. The command required to generate an output set of modified transcripts from transcripts within the input base set that range in length from 300 to 5000, and where 30% of the input transcripts within this range become chimeric (using window length of 250, a maximum of 10 windows and general divergence within windows of 0.4) would be: java -jar ChimSim.jar -ref_set path-to-ref-set.fasta.gz -out_dir path-to-out-dir -min_tln 300 -max_tln 5000 -chim 0.3 -tag -win_ln 250 -max_wins 10 -wgen_div 0.4. There are other ways in which a transcript can be made chimeric; for example, instead of a fixed window size, variable lengths could be used. Here the three categories that are most relevant to graph-based de novo assembly were included, but future developments may include increasing the number of categories available and allowing the user to select which they wish to apply.
A base set of 26,680 transcripts containing all sequences ranging in length of between 300 and 5000 nt present within the fruit fly cDNA library (release-100 from Ensembl85) was created. This base set was used as a reference for simulating reads as required within subsequent sections as well as for the creation of modified base sets containing varying portions of chimerism. This base set along with the original cDNA library from which it was generated, as well as datasets used in subsequent sections describing differential expression analysis and contig creation have been provided as underlying data.88
Chimerism and read mapping
To explore the effects of chimerism on read mapping, the base set was used to create modified sets harbouring varying portions of chimerism. The portions of chimerism introduced ranged from 0% to 95% in steps of five. At each increment, ten replicates were performed. For each replicate five million read-pairs were simulated off the original base set and these were mapped to the modified set generated for that replicate. Mapping was performed with default parameters using Bowtie2,89 following which per-transcript read counts were obtained using the pileup.sh script of the bbmap package.90 R-squared value summarizing the correlation between mapped read count and transcript length were calculated using the R-package (version 4.1.1).91 Additionally, for each replicate of each increment, the total number of successfully mapped reads was recorded. Reads were of length 150 and simulated using CSReadGen (V0.1)92 in a similar manner to that described by Linheiro R & Archer J 2021,40 i.e., where insert size was 300 and no read error, background count variation (above normalized even coverage across all transcripts) or sequence divergence from the reference set was introduced. The approximate per site coverage provided by five million read-pairs across the base set was 26X. To compare the effect of chimerism on read mapping to that of random divergence between reads and the reference set being mapped to, the above was repeated but instead of introducing chimerism during iterations, ChimSim was used to introduce divergence using the -gen_div parameter. Random variation was increased from 0% to 25% in steps of one. For each of the ten replicates associated with a specified level of divergence the total number of reads mapped was counted.
Chimerism and differential expression
To explore the effects of chimerism on the detection of differentially expressed transcripts ten read datasets, each consisting of five million read-pairs, were simulated from the base set (See underlying data).88 These were allocated into conditions labelled as A and B. For the five datasets simulated for condition B, 1000 transcripts were selected at random, using the -rnd_ovr_exp parameter of CSReadGen, to have the required number of reads augmented by a factor of between one and five above the number needed to produce an even coverage across to all transcripts. For all transcripts the required number of read-pairs to produce an even coverage was allowed to vary by a factor of between 0.0 and 0.3, thus providing a level of background variation. Differential expression analysis was performed by obtaining per-transcript count values relative to the base set for each of the ten read datasets, as described in 2.1, and using these counts, in conjunction with their associated condition, as input for DESeq2.76 Within the latter the threshold for the identification of differentially expressed transcripts was a p-adjusted value of 0.05. Differential expression analysis was then repeated iteratively, where during each iteration ChimSim was used to create a modified base set within which a portion of the transcripts present were made chimeric. Reads were mapped to the modified set, counts obtained and DESeq2 employed as before. The portions of chimerism introduced ranged from 5% to 95% chimeric in steps of five. For each level of chimerism the number of differentially expressed transcripts identified between conditions A and B, were compared to these identified when using the unmodified base set for mapping. In addition, the overall presence of the 1000 transcripts specifically marked to have a higher possibility of being over-expressed was monitored as the level of chimerism increased.
Chimerism and de novo assembled contigs
To estimate the extent of chimerisim within de novo assembled contigs, the background relationship between the base set and modified sets, containing incrementing levels of chimerism, was characterized (See underlying data)88). The portions of chimerism introduced went from 5% to 95% in steps of five. Ten replicates were performed at each increment. For each replicate the megablast option86 of the BLAST+ package93 was used to identify the top match within the modified set to each transcript within the base set, and R-squared values summarizing the correlation between alignment length versus transcript length were calculated using the R-package (version 4.1.1). For each transcript within the base set the top ten hits were examined and the one with the longest continuous aligned region used. The distribution of these r-squared values across the incrementing levels of chimerism provided the background expectation for the metric of alignment length versus base set transcript length. Ten million read-pairs were then simulated off the base set, and CStone (v0.01),40 Trinity (v2.12.0)63 and rnaSPAdes (v3.11.1)64 were each used to assemble contigs. In a similar manner to before, megablast was used to compare base set transcripts, from which the reads were simulated, back to contigs and the R-package was used to calculate r-squared values. This procedure of simulating read-pairs from the base set, assembling them and calculating the r-squared values was repeated ten times. Values produced were compared to those calculated for the background relationship across incrementing levels of chimerism. In relation to real data, and as described by Linheiro R et al.,40 two adult fruit fly whole-body samples, from Pang et al. (2021) study on alternative splicing,87 were downloaded from NCBI SRA, study no. SRP297872; run number SRR13251053 for adult 1 and run no. SRR13251054 for adult 2. Reads were 100 nt in length and had been sequenced on Illumina’s Hi-Seq 2000 sequencer (See Underlying data88). Following quality filtering, using Trimmomatic (LEADING:10 TRAILING:10 SLIDINGWINDOW:4:15 MINLEN:36 ILLUMINACLIP:TruSeq3-PE.fa:2:30:10),34 they consisted of 31,543,384 and 29,812,987 read pairs. These were assembled using the three assemblers and megablast was used to compare transcripts from within the fruit cDNA library to the contigs produced. In this case the complete cDNA library was used and not the base set described for the simulations as sequenced reads could represent any transcript present, and not just those from which reads were simulated. As before, the R-package was used to calculate r-squared values summarizing the correlation between alignment length versus transcript length.
For a single replicate, Figure 1 depicts the relationship between transcript lengths and read counts once reads were mapped to the base set, containing 0% introduced chimerism, from which they were simulated. The inset table contains the r-squared value calculated for the line of best fit, as well as those obtained for the other nine replicates. The high values confirm that reads were simulated as expected when specifying even transcript coverage, no background variation, and no read error. When reads simulated in this manner are mapped to modified base sets containing incrementing levels of chimerism (Figure 2A), a progressive lowering of the r-squared values occurs. However, at 95% chimerism a strong correlation remains, the lowest value from ten replicates being 0.8141. R-squared values calculated for just the chimeric transcripts within each set (Figure 2B) are within the range of 0.8158 to 0.8638 across all increments. This is consistently lower than the values calculated for the non-chimeric transcripts (Figure 2C) which indicates that when all transcripts are present, it is the presence of the chimeric ones that lowers the overall values. When r-squared values obtained using transcripts associated with each individual category of chimerism are plotted (Figures 3A to C), the over-extension category had less of an effect than those of windowed variation and window shuffling. In relation to the overall number of reads mapped (Figure 4A), although mapping sucess decreases as chimerism is increased, the lowest point occurs for a replicate at 95% chimerism, when 77% of reads were still mapped. The combination of these results indicated that, even when faced with extreme chimerism, on a surface level read mapping does not appear to perform poorly when looking at basic count values. This is because much of the variation introduced by chimerism is not novel and the majority of reads will find a transcript to map to. Importantly, this suggests that there could be a hidden impact of chimerism on downstream data analysis that is hard to predict based on mapped read counts alone. Figure 4B indicates that this is not the case for the introduction of random variation between the reference set and the reads being mapped, where a rapid decline in mapping success is evident.
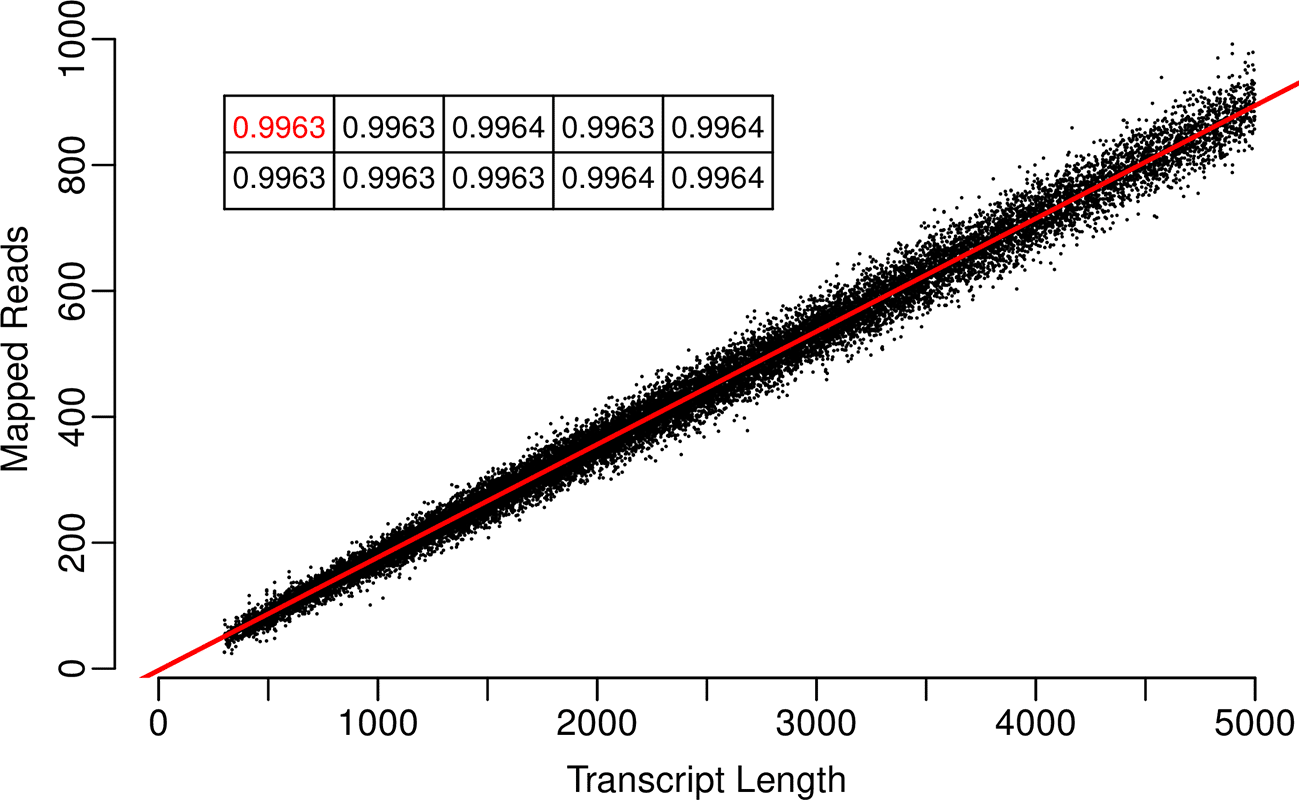
Reads containing no sequencing error, and distributed evenly across all transcripts, were mapped to the base set (containing 0% introduced chimerism) from which they were simulated. The x-axis indicates the transcript length, and the y-axis indicates the number of mapped reads. The r-squared value associated with the line of best fit is indicated within the inset table (in red). The other r-squared values within the table indicate those associated with the other nine repetitions of read simulation and subsequent mapping to the base set.
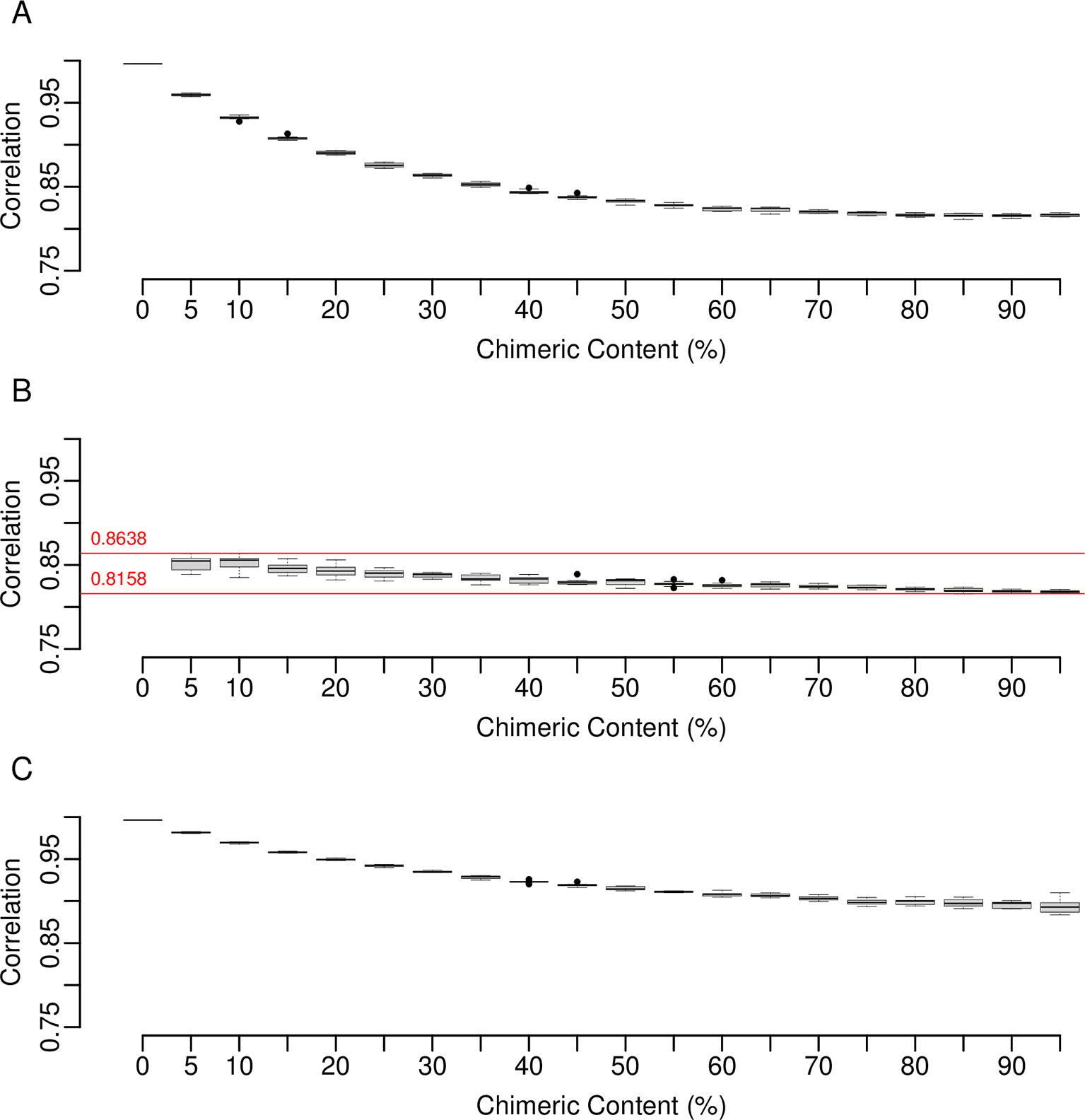
(A) R-squared values (y-axis) summarizing the correlation between transcript length and mapped read count in relation to reads simulated off the base set but that are mapped to related sets containing incrementing levels of chimerism (x-axis). At each increment of ten replicates were performed. (B) Same as for (A) but only the chimeric transcripts, at the indicated increment of chimerism, within the modified set are included. (C) Same as for (A) but only the non-chimeric transcripts, at the indicated increment of chimerism, within the modified set are included. In all cases, the medians are shown within each box. Whiskers extend to the furthest data point that is within 1.5 times the interquartile range and points beyond this are outliers (black circles).
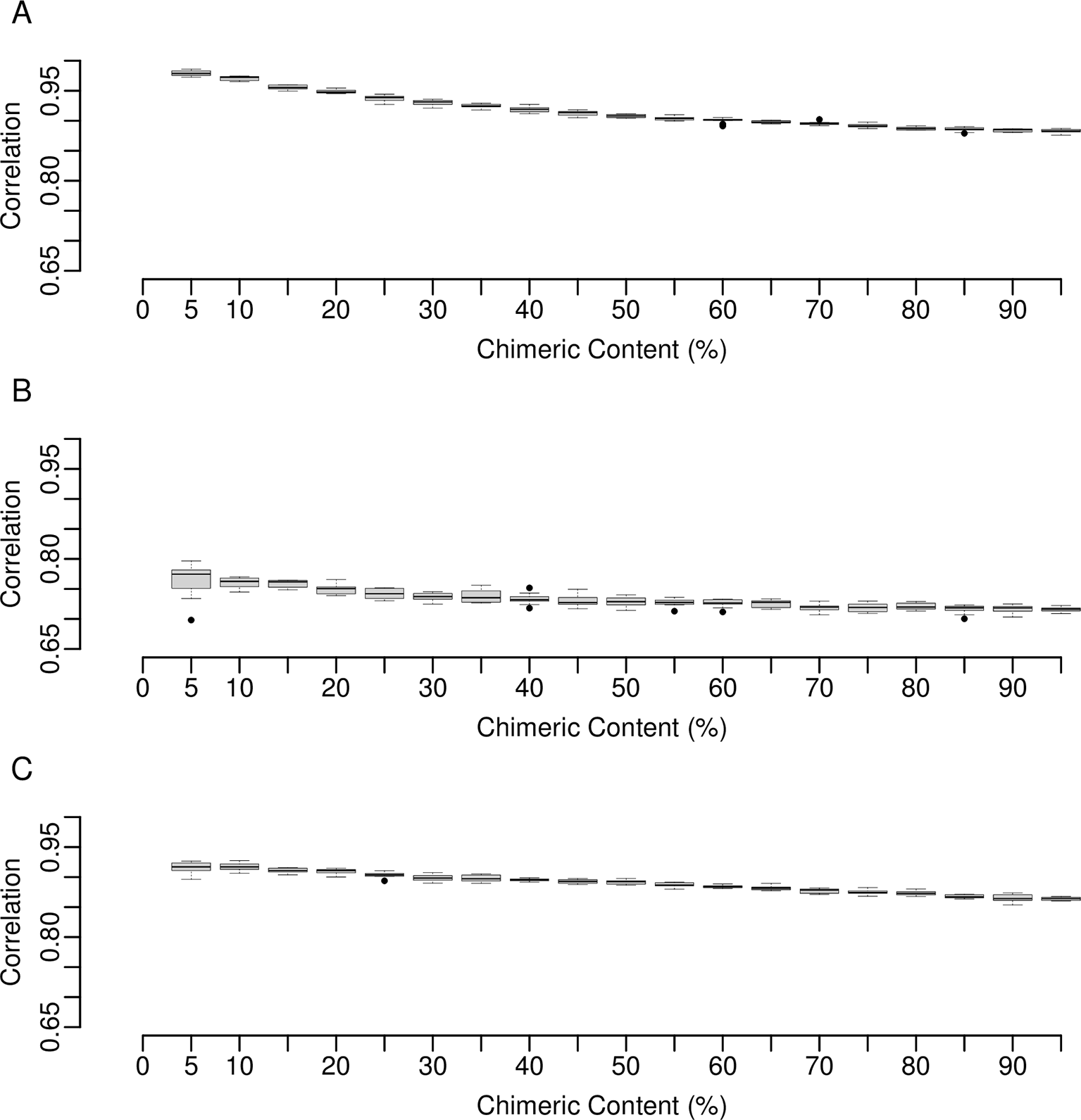
R-squared values (y-axis) between the length of individual transcripts (to which chimerism had been introduced) and mapped read count, i.e., those presented in Figure 2B, were divided into the three categories of chimerim implemented within ChimSim. These were: (A) Over-extension, (B) Window variation and (C) Window shuffling. In all cases the medians are shown within each box. Whiskers extend to the furthest data point that is within 1.5 times the inter quartilerange and points beyond this are outliers (black circles).
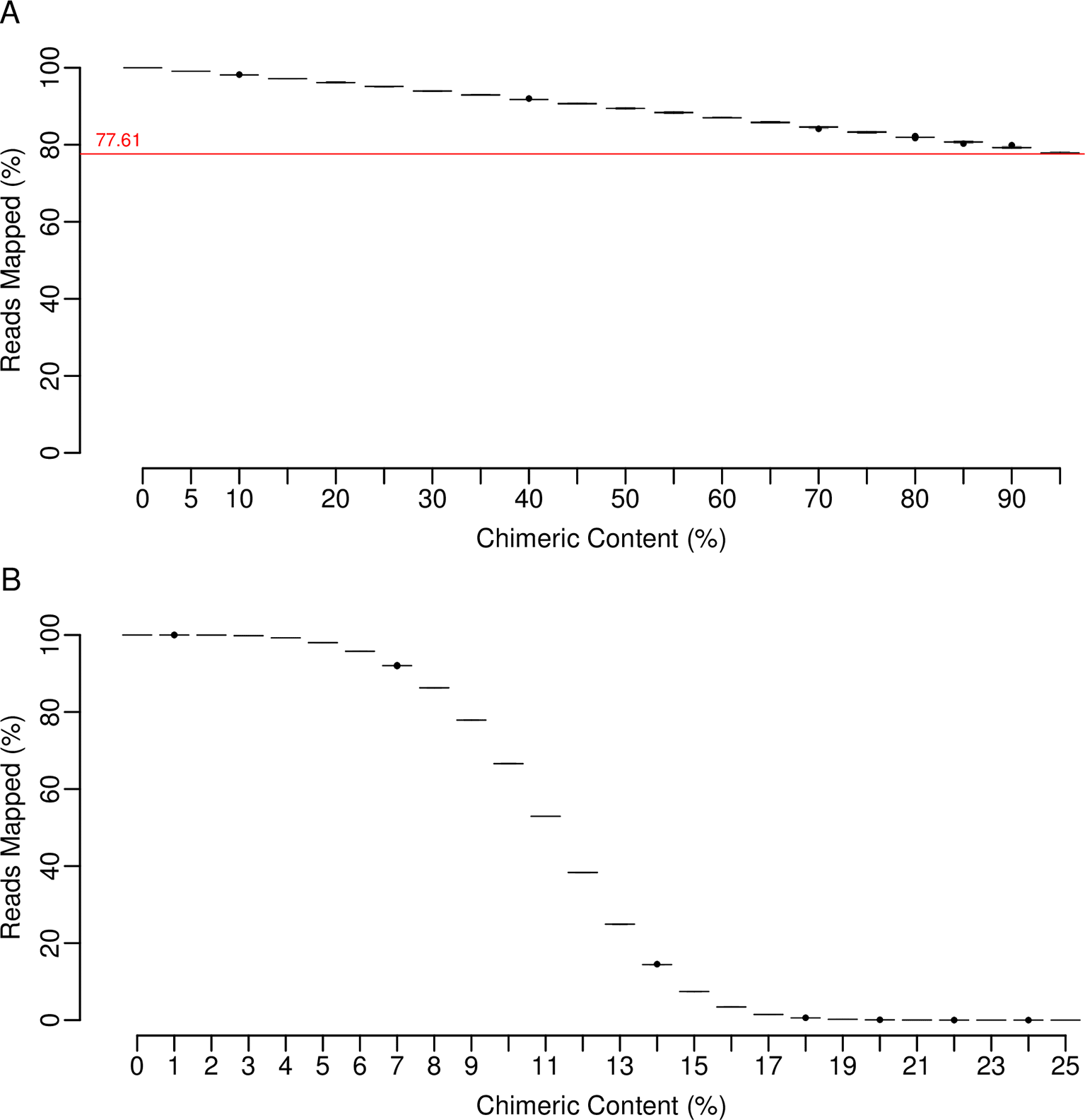
Reads simulated off the base set were mapped to: (A) modified base sets containing incrementing levels of chimerism (x-axis) following which the total number of reads successfully mapped counted (y-axis) and (B) modified base sets containing incrementing levels of sequence divergence (x-axis) and the total number of reads successfully mapped counted (y-axis). In panel (A) the red line indicates the lowest mapped read percentage achieved across all replicates and increments. In all cases the medians are shown within each box. Whiskers extend to the furthest data point that is within 1.5 times the interquartile range and points beyond this are outliers (black circles).
In Table 1 it is observed that when datasets belonging to conditions A and B were mapped to the base set containing 0% introduced chimersim, and differential expression analysis performed, 2853 and 400 transcripts were identified as being over- and under-expressed. Of the 2853 over-expressed transcripts, 980 were within the set of 1000 transcripts randomly selected for increased representation within condition B. The remaining 1873 being a consequence of the random background variation applied. The other rows of Table 1 show that despite chimera’s having a relatively low effect on general mapping success, i.e., Figures 2 and 4, increasing the level of chimerism within the base set prior to mapping and subsequent differential expression analysis has a large effect on the identification of differentially expressed transcripts. Of the 980 transcripts that were identified as being over-expressed, that belonged to the set of 1000 transcripts selected increased read representation within condition B, the number detected at each incrementing level of chimerism rapidly diminished. Likewise, but more generally, Figure 5 indicates that for each 5% increment in chimerism, the number of overall transcripts detected as being over- and under-expressed that agree with those identified in the absence of chimerism (Table 1 - row 1) also diminishes. This highlights the ambiguity that can be introduced during downstream data analysis as a result read mapping yielding unreliable, not necessarily diminished, counts when faced with chimerism.
The red text indicated row one, the numbers obtained when using a reference set containing no introduced chimerism. The last column indicates the number of transcripts identified as being over-expressed that were within the set containing the 1000 transcripts randomly selected for increased read representation within condition B during read simulations, i.e., increased chance of over-expression.
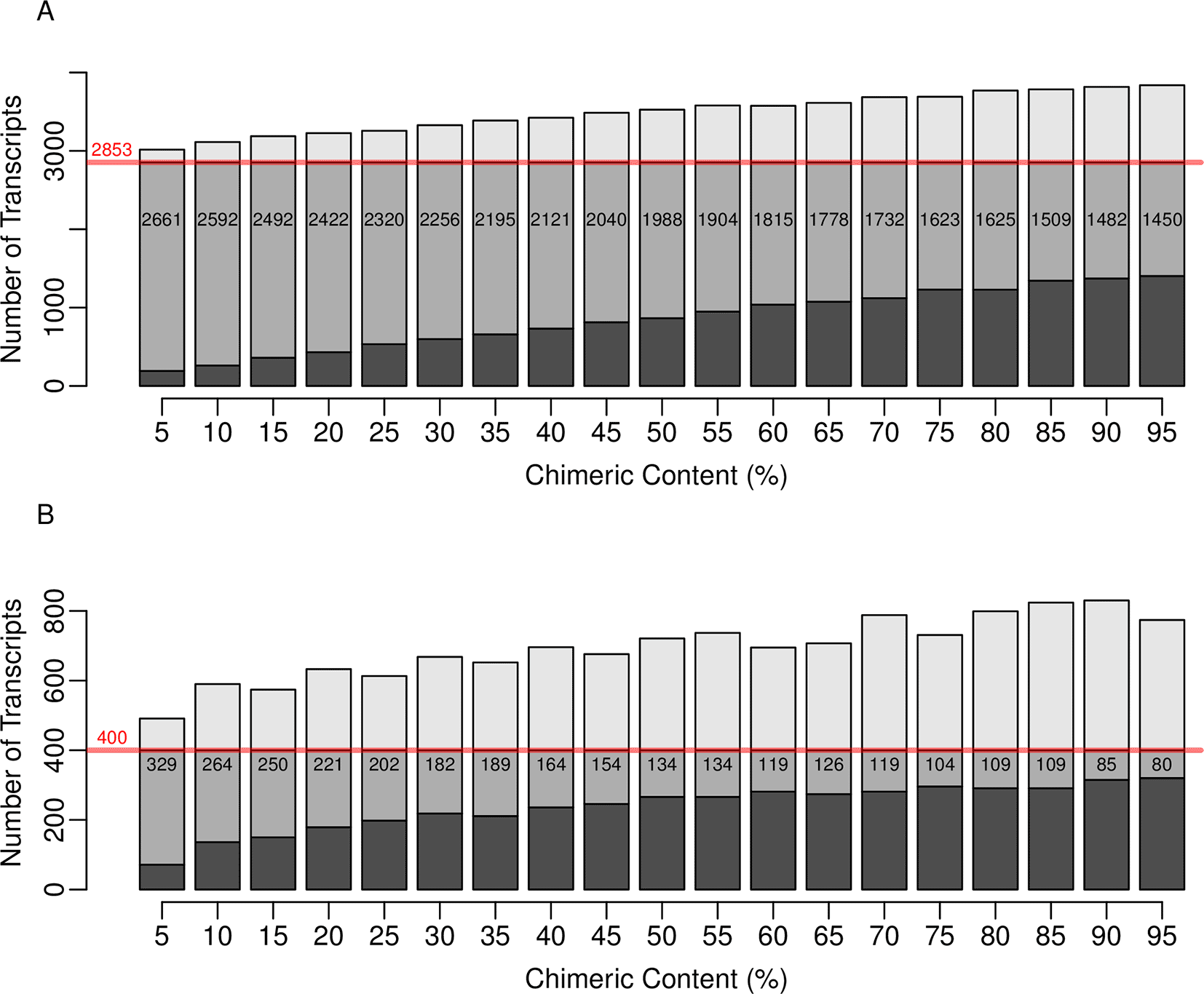
Ten paired-read datasets were simulated and divided evenly into two conditions. Unlike previous simulations, per-transcript read representation was allowed to vary. Additionally, within one of the conditions, 1000 transcripts were over-represented across the five replicates. Differential expression analysis between the two conditions was performed, using the non-chimeric base set, in order to obtain a list of over- and under-expressed transcripts. Differentially expression was then iteratively repeated in a similar manner, but where the extent of chimerism within the reference set used was incremented (x-axis). The middle grey section of each bar represents the number of (A) over- and (B) under-expressed transcripts identified at the indicated level that were also identified when performing differential expression analysis using the non-chimeric base set. In both panels the dark grey area of each bar indicates transcripts that were identified as being differentially expressed solely when using the non-chimeric reference set. The corresponding light grey bar represents transcripts identified as being differentially expressed when solely using a modified reference into which the indicated level of chimerism was introduced. The red lines in both panels indicate the total number of over- and under- expressed transcripts identified when using the non-chimeric base set.
For the three assemblers, Figure 6A displays the r-squared values that describe the correlation between alignment lengths and contig lengths. The alignments used were those between base set transcripts and best matching contigs. Higher values indicate that larger portions of the contigs were aligned, thus indicating better assemblies. The values obtained, ranging from 0.8832 to 0.9591, suggest that as a whole contigs produced by all assemblers reflected well the regions of the base set transcripts to which they aligned. Figure 6B shows the distribution of r-squared values describing the equivalent correlation but, instead of assembled contigs, modified base sets of transcripts containing varying levels of chimerism were used. Direct comparison to the equivalent values obtained for the three assemblers (Figure 6A) suggests that the level of chimerism within the assembled contigs could be in the range of between 5-15%. Figure 6C shows that the r-squared values obtained for contigs assembled from RNA-Seq data obtained from the two fruit fly whole adult samples are only slightly lower than those from the simulated datasets and thus the expected level of chimerism would not be dissimilar. Given the effects that such sequences have on differential expression analysis, even at the 5-15% levels (Table 1 and Figure 5), the analysis performed for Figure 6 highlights the need to either quantify these sequences further during the de novo assembly process or to circumvent the problem of chimerism completely by moving towards approaches that utilize long read technologies.

(A) For the contigs created by each assembler, across each of ten replicates, r-squared values describing the correlation between alignment length and contig length were calculated. The alignments used were those between the top contig match for each transcript within the base set from which reads were simulated. The red dotted lines indicate the maximum and minimum value obtained. (B) The distribution of r-squared values describing the equivalent correlation described in (A), but instead of assembled contigs, modified base sets of transcripts containing varying levels of chimerism were used. The dotted red line indicates where the maximum and minimum values obtained for the three assemblers indicated within panel A could be projected on-to the x-axis. (C) Following quality filtering of two adult fruit fly whole-body samples from Pang et al. (2021),40 consisting of 31,543,384 and 29,812,987 read pairs, reads were assembled using each of the three assemblers and the r-squared values described for (A) calculated. In all cases the medians are shown within each box. Whiskers extend to the furthest data point that is within 1.5 times the interquartile range, and points beyond this are outliers (black circles).
Figure 7A indicates the range of transcript lengths present within the modified base set created across each replicate used in Figure 6B. The slight increase observed with incrementing chimerism is as a result of the transcripts selected for over-extension. Despite this minimal increase in the overall length distributions, Figure 7B indicates that the number of base set transcripts being represented by a match within the modified sets is rapidly reduced with incrementing chimerism. When 5% of the transcripts within the modified sets are chimeric the median number of transcripts within the base set finding a megablast match is 22495, whilst at the 15% level of chimerism it is 194343. This is due to sequence variation associated with chimerism increasing. Although preliminary, this suggests that the extent of chimerism within de novo assembled contigs will also have an effect on annotation tools that rely on searches based on sequence similarity.

(A) Lengths (y-axis) of transcripts within the modified base sets following the introduction of chimerism to varying degrees (x-axis). The numbers along the top of each box and whisker indicate the number of transcripts above 5000 nt in length (red line). (B) The number of base set transcripts (y-axis) finding a representative match within the modified base sets containing incrementing degrees of chimerism. Despite the consistency in modified base set transcripts lengths as chimerism is introduced (panel A), the number of base set transcripts represented rapidly diminishes. In all cases the medians are shown within each box. Whiskers extend to the furthest data point that is within 1.5 times the interquartile range and points beyond this are outliers (black circles).
Although it is known that the de novo assembly of short-read RNA-Seq data can result in chimeric contigs, the extent of such chimerism has been poorly quantified, as has the effects that such chimerism has on data analysis. In this study we have demonstrated these effects on read mapping and on the identification of differentially expressed transcripts. We have also indicated to what extent such chimerism could be expected within contigs assembled using three graph-based assembly tools. Despite all tools performing well, the rapid consequence of even low levels of chimerim on results interpretation, indicate that further effort is required to include information relevant to chimera quantification, and that results dependent on short-read assembly must be present within the context of this information. An inability to make this improvement to current assemblers would suggest that transcriptomics experiments must strive to move away from using short-read data. If not the consequences on scientific robustness in relation to results-base conclusion will be difficult to mask.
Zenodo: Quantification of the effects of chimerism: datasets
This project contains the following underlying data:
BaseSetTranscripts.zip: Contains a file with all transcripts present within the Ensembl release-100 of the fruit fly cDNA library and another file containing sequences ranging in length of between 300 and 5000 nt from that cDNA library
DEReads.zip: Contains the ten sets of paired reads, divided into conditions A and B as indicated, that were used for differential expression analysis.
DEChimSimRefs.zip: contains the references sets harbouring varying levels of chimerism that were used for differential expression analysis.
DeNovoAssemblies_SimulatedData.zip: Contains the de novo assemblies.
Reads_RealData_WholeBody_1.zip: Contains the whole body read datasets from adult fruit fly 1 following quality filtering.
Reads_RealData_WholeBody_2.zip: Contains the whole body read datasets from adult fruit fly 2 following quality filtering.
DeNovoAssemblies_RealData.zip: Contains the assemblies generated when using the previous two datasets as input.
All data is available under the terms of the Creative Commons Attribution 4.0 International license
John Archer: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – original draft. Raquel Linheiro: Visualization. Raquel Linheiro and John Archer: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – review & editing.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)