Keywords
Polyphenols, mulberroside A, molecular docking, cancer, molecular dynamic simulation, protein kinase inhibitors.
Polyphenols, mulberroside A, molecular docking, cancer, molecular dynamic simulation, protein kinase inhibitors.
Human epidermal receptors (HERs) have been shown to play a crucial role in a variety of cancer forms, including non-small cell lung cancer (NSCL), which kills more people than prostate cancer, breast, colorectal combined and around 20% of breast cancers, which are characterized by overexpression of human HER2 protein and associated gene (Majeed et al., 2021).
There are four HER receptors: HER1, also known as EGFR, HER2, HER3, and HER4. The members of the HER family are transmembrane receptor tyrosine kinases, which are enzymes that catalyze tyrosine phosphorylation, specifically the transfer of adenosine triphosphate (ATP) to tyrosine residues on protein substrates and the subsequent recruitment and activation of downstream signaling proteins. This results in the activation of downstream signaling pathways that promote cell proliferation, differentiation, migration, survival, adhesion, and angiogenesis. Numerous malignancies have been associated with a mutation that enables some tyrosine kinases to be constitutively active and/or occasionally act improperly.
The extracellular domain (ECD), a transmembrane segment, and an intracellular portion all comprise the structure of HER receptors. The intracellular region consists of a juxtamembrane segment and a functional protein kinase domain (with the exception of HER3, which lacks tyrosine kinase activity and must be activated in conjunction with another family member). The catalytic domain or the tyrosine kinase domain (Figure 1) contains the conserved ATP binding groove, which is required for ATP binding (Jura et al., 2009). The tyrosine kinase domain (TKD) is made up of an activation loop, a C-lobe, and an N-lobe, which contains a C α-helix. The activation loop, which is a polypeptide region outside the binding site cleft that, upon phosphorylation, induces kinase activation and C α-helix must adopt a particular, “active” configuration in order for a TKD to function (Wintheiser & Silberstein, 2021; Adams, 2003).
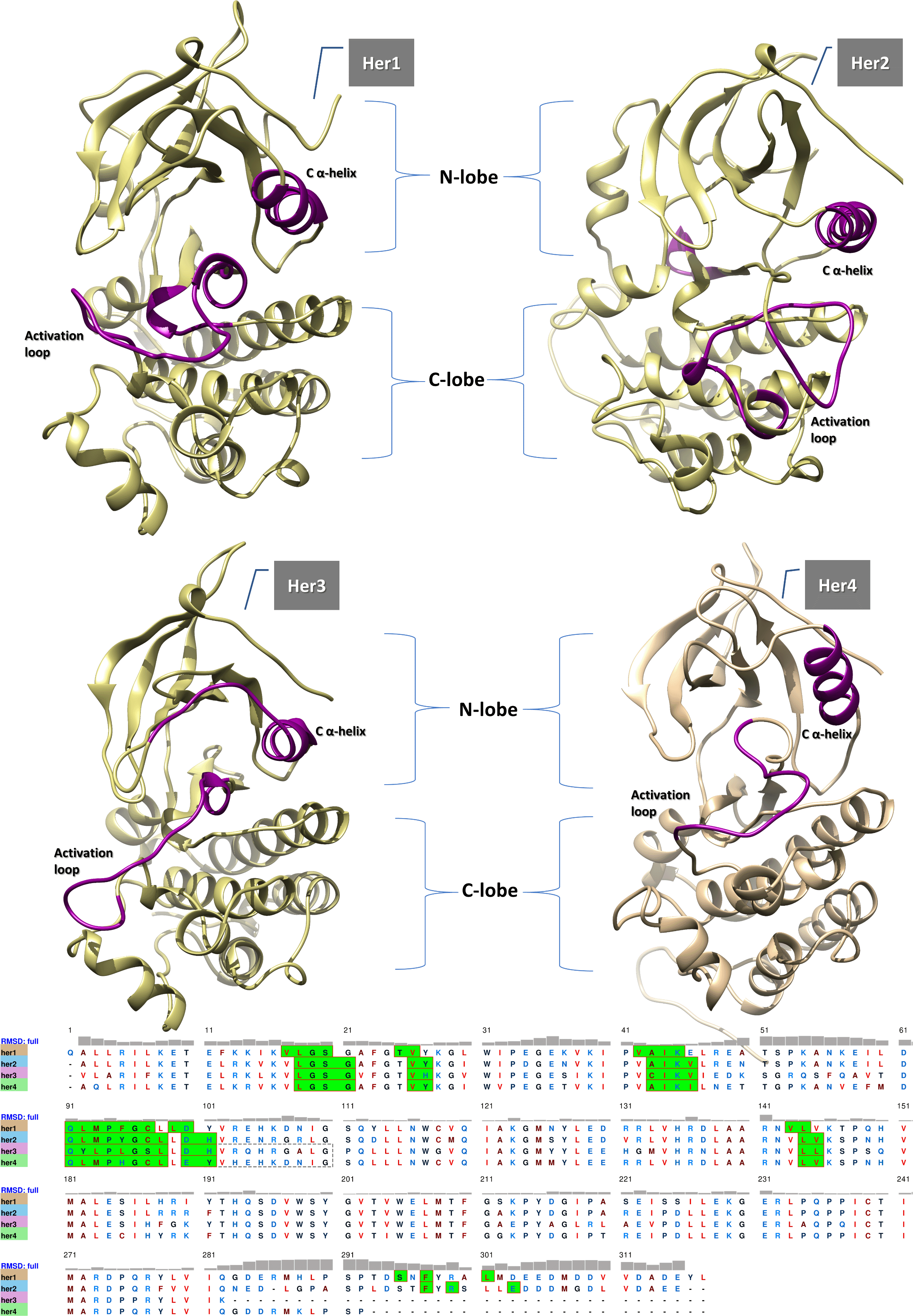
There are two lobes (C-lobe and N-lobe) that are spanned by a loop-like structure called the activation loop (purple in color). The N-lobe contains the C α-helix (purple in color) which is essential for the catalytic activity of the enzymes. The sequence alignment reveals the residues that are primarily responsible for forming the binding site (green-shaded) and that are predicted to interact with mulberroside A, along with their RMSD values, which show the deviation from each other.
The development of acquired resistance to chemotherapy is one of the most perplexing concerns confronting oncologists. Although cancer cells initially respond to chemotherapy, later as treatment progresses, mutations and epigenetic alterations may occur, resulting in a high degree of plasticity of these cells that leads to the emergence of resistance. Due to receptor mutations, anti-HER resistance has risen over the years, necessitating an ongoing search for new inhibitors.
Numerous studies on the antioxidative, anti-inflammatory, and anticarcinogenic properties of dietary polyphenolic substances have been carried out extensively. In this regard, dietary polyphenols have been shown in preclinical models to display chemo-preventive and therapeutic benefits against a variety of malignancies. However, available medications have a high level of unspecific cytotoxicity, which has a detrimental effect on normal cells as well. As a result, the search for anticancer agents derived from natural medicinal plants has progressively increased throughout the years (Mechchate et al., 2021). Phenolic compounds are one of the most abundant and diverse classes of plant metabolites, with over 8,000 compounds described. Stilbenes, benzoquinones, acetophenones, flavonoids, anthocyanins, phenolic acids, catechins/epicatechins, ellagitannins, and proanthocyanidins are the various polyphenolic classes. The most extensively studied of these are (-)-epigallocatechin-3-gallate, resveratrol (found in grape skin), and curcumin (found in turmeric) (Pandey et al., 2009). In this context, certain polyphenols have been shown to act as chemosensitizers, enhancing the efficacy of chemotherapeutic agents when used in combination. For instance, it has been demonstrated that the flavonoid silibinin considerably showed activity against the resistant lung cancer cells to the tyrosine kinase inhibitor (TKI) erlotinib which led to a suggestion of using silibinin in combination with TKIs as a treatment regimen (Verdura et al., 2021). Curcumin is another polyphenol which, when combined with 5-fluorouracil, demonstrated antimetastatic activity (colon carcinoma) (Yang et al., 2017). Additionally, it has been reported that resveratrol enhances the anticancer effects of gemcitabine by reducing the emergence of resistance in pancreatic cancer cells and sensitizing glioma stem cells to radiation (Zhou, et al., 2019). Stilbenes are a non-flavonoid polyphenolic phytochemical with a 1,2-diphenylethylene nucleus. They hold exceptional promise for the prevention and treatment of cancer, due to their antioxidant, cell death activation, and anti-inflammatory properties, all of which are associated with low toxicity in vivo (Sirerol et al., 2016). Numerous methoxy and hydroxy derivatives of trans-stilbene have been shown to inhibit the growth of varied human cancer cells by causing cell apoptosis (Parida et al., 2018). Mulberroside A is a stilbenoid found in Morus alba (the white mulberry) and a diglucoside of oxyresveratrol. Due to its successful docking results, mulberroside A will be the focus of this study, and its effect on the dynamics of the proteins will be investigated further.
The crystal structures of the TKD of the HERs were obtained from the protein data bank. 3POZ (chain A), 3RCD (chain A), 6OP9 (chain A), and 3BBT (chain B) are the PDB codes for HER1 (sequence 701-1017), HER2 (sequence 710-1022), HER3 (sequence 680-960), and HER4 (sequence 683-973), respectively. The missing residues were added by Salilab modeler (Pieper et al., 2006) and they were for HER1 748-754, 734-737, 868-874, 1004-1009 segments, for HER2 757-760, 867-883, 992-999, 1013-1015 segments, for HER3 847-851 segment, and for HER4 844-858 segment. The evaluation of the modeled missing parts was carried out by normalized Discrete Optimized Protein Energy (zDOPE), an atomic distance-dependent statistical score (Shen & Sali, 2006). The residues of the proteins were renumbered as a requirement of the modeler starting from number one (HER1; 1-317, HER2; 1-312, HER3; 1-280, HER4; 1-290). The binding sites have been chosen based on the coexisted inhibitors that occupy an orthosteric site, which is the active one.
Prior to docking, all water molecules, ions, and ligands were eliminated from the enzymes. These enzymes were then prepared for docking by adding hydrogen atoms and charges (Charges were modeled by AMBER ff14SB for the residues) using Dockprep's UCSF chimera-building feature (Pettersen et al., 2004). Afterward, the ligand was obtained from PubChem and prepared for docking by adding hydrogen atoms and charges. Charges were modeled by AM1-BCC (Austin model with bond and charge correction) for the ligand (Maier et al., 2015; Jakalian et al., 2002). The ligand structure was minimized and optimized by MM2 force field that built in chemoffice software (Hocquet & Langgård, 1998).
The docking process and the estimated free binding energy were handled by the AutoDock Vina (Trott & Olson, 2010). Up to 10 poses were produced for the ligand within the binding site of the enzymes, and the selection was for the pose that was characterized by a high binding energy. The docking process was validated (Figure 2) by redocking the native crystallized ligands (HER1; TAK285, HER2; TAK285, HER3; bosutinib, HER4; lapatininb), and the webserver DockRMSD was used to calculate the RMSD between the redocked and crystallized ligands (Bell & Zhang, 2019). The box grid dimensions were determined based on the production of the best proximity to the conformation of the inhibitors from the original x-ray crystal structure. The dimensions were as follows: HER1 (Center |18.544|34.556|4.399, size|37.219|19.332|32.395), HER2 (Center |8.879|1.626|24.569, Size |39.994|37.149|22.890), HER3 (Center|-46.127| 19.229 |21.339, Size |26.413|19.229|21.339), HER4 (center|-39.845|49.482|21.561, Size|39.904|33.612|32.051).
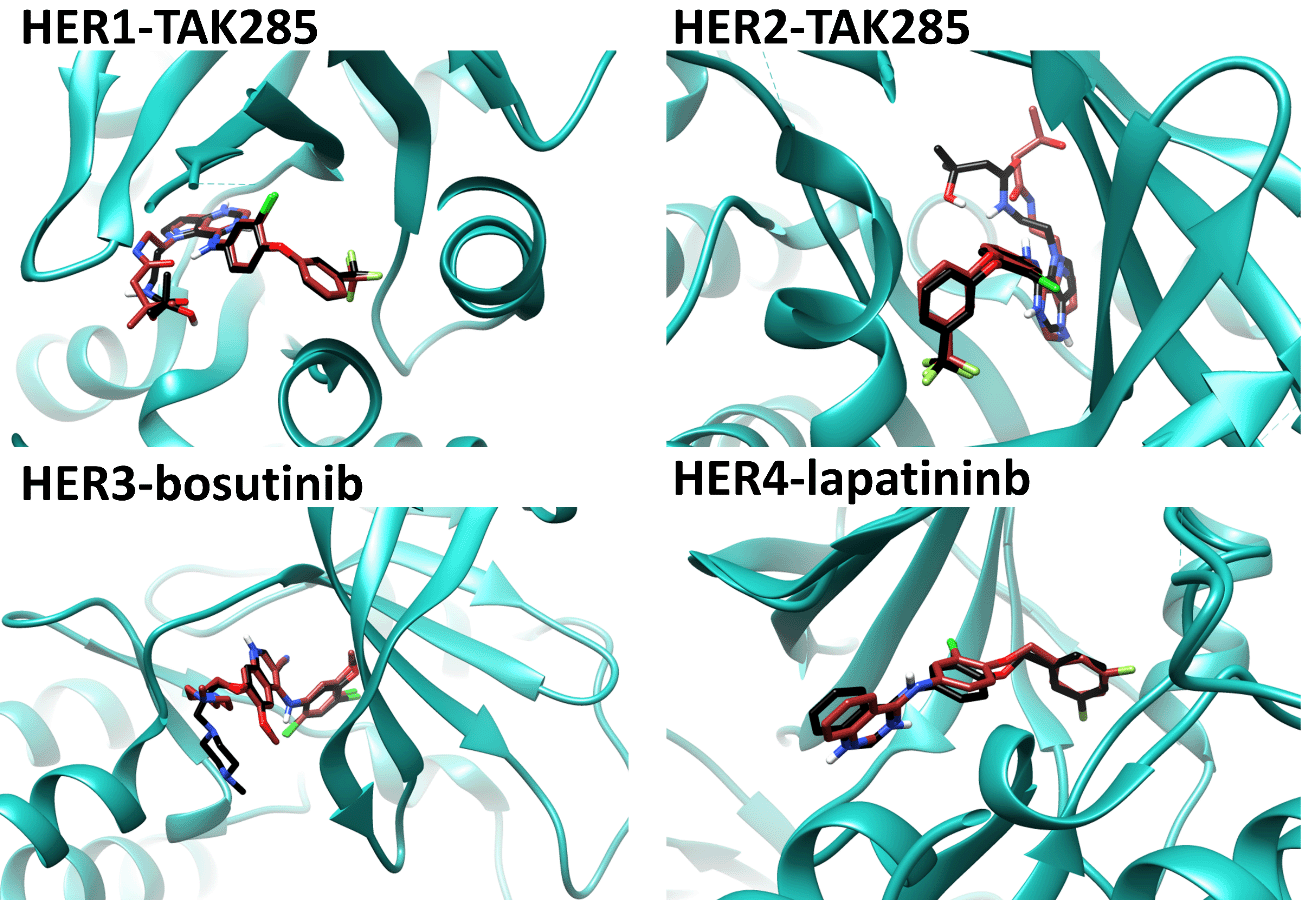
The RMSD values indicating the difference between crystallized and redocked ligands for HER1, HER2, HER3, and HER4 are 1.125, 2.036, 2.515, and 1.341, respectively. The redocked native ligands have docking scores of -44.35, -41, -35, and -43.5 kJ/mol for TAK285 (HER1 and HER2) bosutinib (HER3), and lapatininb (HER4), respectively.
The MD simulation process was carried out for the ligand-protein complexes that had given the best-free binding energy scores. The GROMACS 2018.2 package (Abraham et al., 2015) was used along with Charmm27 to parametrize the residues (Brooks et al., 2009). The system was solvated by applying the TIP3P water model (Jorgensen et al., 1983). Swissparm server (http://www.swissparam.ch) were employed to generate the topology and parameters of mulberroside A. A triclinic periodic box (periodic boundary condition) was generated, and the distance between the edge of the box and the complex of protein-ligand was set to 1.0 nm (Fletcher & Powell, 1963). Chloride and sodium ions were added at a concentration of 0.15 M to achieve a neutral system. Energy minimization was run using the steepest descent (Jaidhan et al., 2014) to make the system's potential energy reach -7×105, -5×105, -6×105, and -6×105 kJ/mol for HER1, HER2, HER3, and HER4, respectively. The energy step size was set to 0.01 nm, and a maximum of 50,000 steps was introduced. The temperature represented by the isochoric-isothermal (NVT) ensemble was equilibrated to 310.15 K. The time of equilibration was fixed to 1 ns with a time step of 2 fs. Afterward, isothermal-isobaric (NPT) equilibration was processed for 1 ns with a time step of 2 fs. Further settings of the NPT ensemble were 0.1 nm for both van der waals (VDW) and electrostatic interactions. The modified Berendsen thermostat for temperature coupling (310.15 K), the particle mesh ewald (PME) for calculating the long-range electrostatics, and the Parrinello-Rahman method as a barostat for pressure coupling were also used (Parrinello & Rahman, 1981). The MD simulations were performed for 200 ns with the same settings mentioned for NPT. The free binding energy, VDW interactions, electrostatic interactions, SASA, and the polar solvation energy were carried by the MM/PBSA method (Genheden & Ryde, 2015). RMSD, RMSF, Rg, and the PCA tools that are built-in gromacs software were used to monitor the conformations of the proteins. (Fuglebakk et al., 2012; Lobanov et al., 2008; Balsera et al., 1996). For additional information on the MD simulation parameter, a link is provided in the section of data availability.
Concerning the modeling of the missing parts of the receptor kinases, the zDOPE scores for HER1, HER2, HER3, and HER4 are -1.84, -1.94, -1.87, and -1.58, respectively; negative values indicate superior models. Upon docking mulberroside A into these domains, all displayed a high binding affinity; however, HER1 and HER2 displayed the highest affinity, with scores of -40.54 and -40.12 kJ/mol, respectively (Table 1). Compared to the docking scores of the co-crystallized native ligands (mentioned in the caption of Figure 2), the binding energy of mulberroside is very close.
As shown in Figure 4, five H-bonds were formed between the residues Ser20, Leu77, Met93, Asp155, and Phe156 of HER1 and the ligand. The bonds that were established with Met93 and Phe156 were the strongest of the five, and these bonds remained within the range of H-bond length (which is 2.5-3.5 Å) throughout the entirety of the simulation. HER2 formed four H-bonds by the residues Ser74, Leu76, Leu87, and Asp154. The H-bond formed by Leu87 is the most stable of the four because it did not exceed the length threshold for a strong H-bond while the simulation was being run. The ligand was unsuccessful to build strong H-bonds with HER3; upon docking, the two H-bonds were perfectly with good length (2.57 Å with Leu92 and 2.39 Å with Ser96) but unfortunately, did not survive the MD simulation. The five H-bonds that were recognized within the binding site of HER4 upon the binding of the ligand were with Thr89, Glu99, Arg140, and Asp154 residues. The strongest were the bonds with Asp154 and Glu99, which remained stable during the simulation.
Molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) calculation was performed for the last 100 frames (the last 1 ns) and the results revealed some relevant outcomes to the docking score except for the interaction with HER2, which was scored -52.16 kJ/mol, the lowest due to low electrostatic interaction (energy) and a high wasting of binding energy in polar solvation energy (Table 2). The binding energy with HER1 is -119.1 kJ/mol, and it is based on strong electrostatic and VDW forces. Even though binding to HER4 required a high polar solvation energy, there was a noticeable offset from the energy gained from VDW, electrostatic, and SASA interactions.
Figure 3 depicts the binding poses of mulberroside A within the four kinases. In the case of HER1, mulberroside A was close enough to interact with both the activation loop (via VDW interaction with Thr154 and two H-bonds with Asp155 and Phe156) and the C α-helix (via VDW interaction with Met65). In HER2, the ligand has successfully interacted through VDW with Met65 of the C α-helix while no direct interaction with the activation loop was observed. In HER3, the ligand was unable to successfully establish direct interaction with the C α-helix; however, Val157 of the activation loop was close enough to the ligand to engage in direct VDW interaction. Concerning HER4, no interaction was found between the activation loop and the ligand. On the other hand, VDW interaction was found with the C α-helix, which was mediated by the residue Met65.
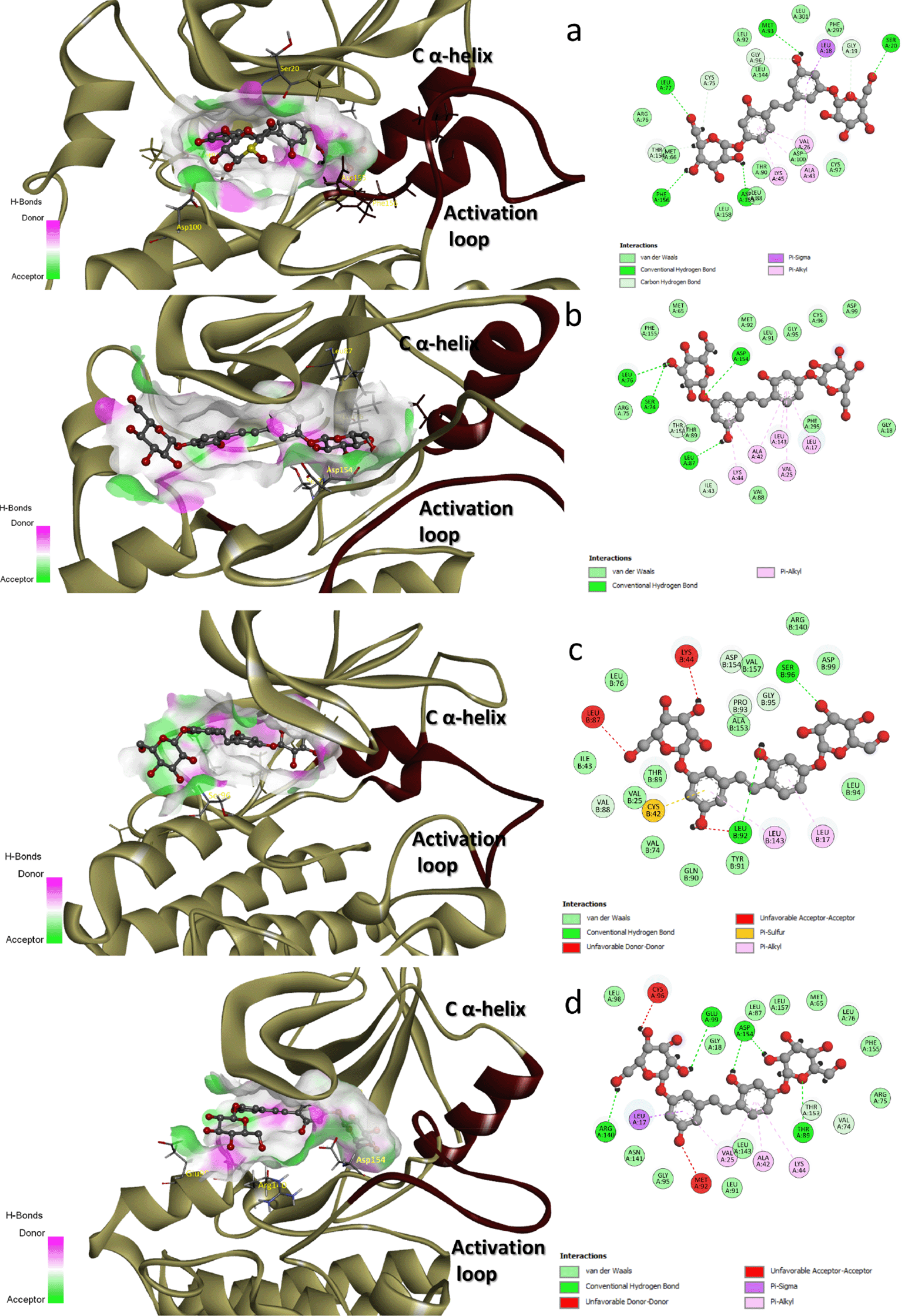
There were roughly 10 H-bond donors and 11 H-bond acceptor spots in the binding pocket of HER1. 10 donors and 8 acceptors made up HER2. There were 10 donors and 12 acceptors for HER3. 8 donors and 10 acceptors comprise HER4. This led us to deduce that an increase in acceptors within the binding site would have an impact on the binding affinity of mulberroside A. By modifying the responsible moieties, the binding affinity of the ligand could be increased by avoiding clashes (acceptor-acceptor clashes) with Lys44 and Leu87 in the HER3 binding site and with Cys96 and Met92 in the HER4 binding site.
For MD simulation, the RMSD plot (Figure 5a) over the duration of the simulation showed that the conformation of the TKDs had undergone a very slight shift. When the ligand bound to the TKD of HER1, the protein became more rigid, resulting in less conformational variation than when the enzyme was in its apo state. This conformational variability is caused by one of the terminals in the apo form fluctuating more, as demonstrated by the region 1—10 in the RMSF plot (Figure 5b). Whereas in holo TKD of HER2, region 150—200 was the most variable segment, representing a portion of the binding site and extending into the activation loop. Both the apo and holo TKD of HER2 exhibited behavior that is, to some extent, comparable. Both of them fluctuated more at the beginning of the simulation, but by 90 ns, they had reached a point of convergence. The region 40—60, which included the binding site and a portion of the C -helix, shared the same overtones as the region 150—200. When compared to HER1 and HER2, the TKD of HER3 appeared to have a greater degree of instability in regard to regions 40—60 (C α-helix region) and 150—200 (activation loop region). The apo form and the holo form had the same tone for region 150—200, but the apo form has a different tone for region 40—60 due to the fact that the apo form was more flexible. The last few nanoseconds of simulation could reveal that holo-HER3 became more stable and less prone to conformational variations than apo-HER3. The high RMSD records were the direct result of the extreme fluctuation that occurs at one of the terminals in the TKD of HER4. In the apo form, the fluctuation began at 50 ns, whereas in the holo form, it began at 170 ns. In general, the apo form is very dynamically active in the region of the terminal and the region 150—200, whereas the holo form is characterized by a calm motion to some extent, in contrast to the apo form.
All of the TKD receptors showed more compactness in unoccupied form rather than occupied form as shown in the radius of gyration plot (Figure 5c), the most noticeable being HER4 which upon binding of the ligand led to a decrease in the compactness by 2 Å which was the largest. The solvent accessible surface area calculation (Figure 5d) showed an expansion (unfolding) in the structures of HER1 and HER4 in the presence of the ligand comparable of its absence, without any remarkable change in HER2 and HER3.
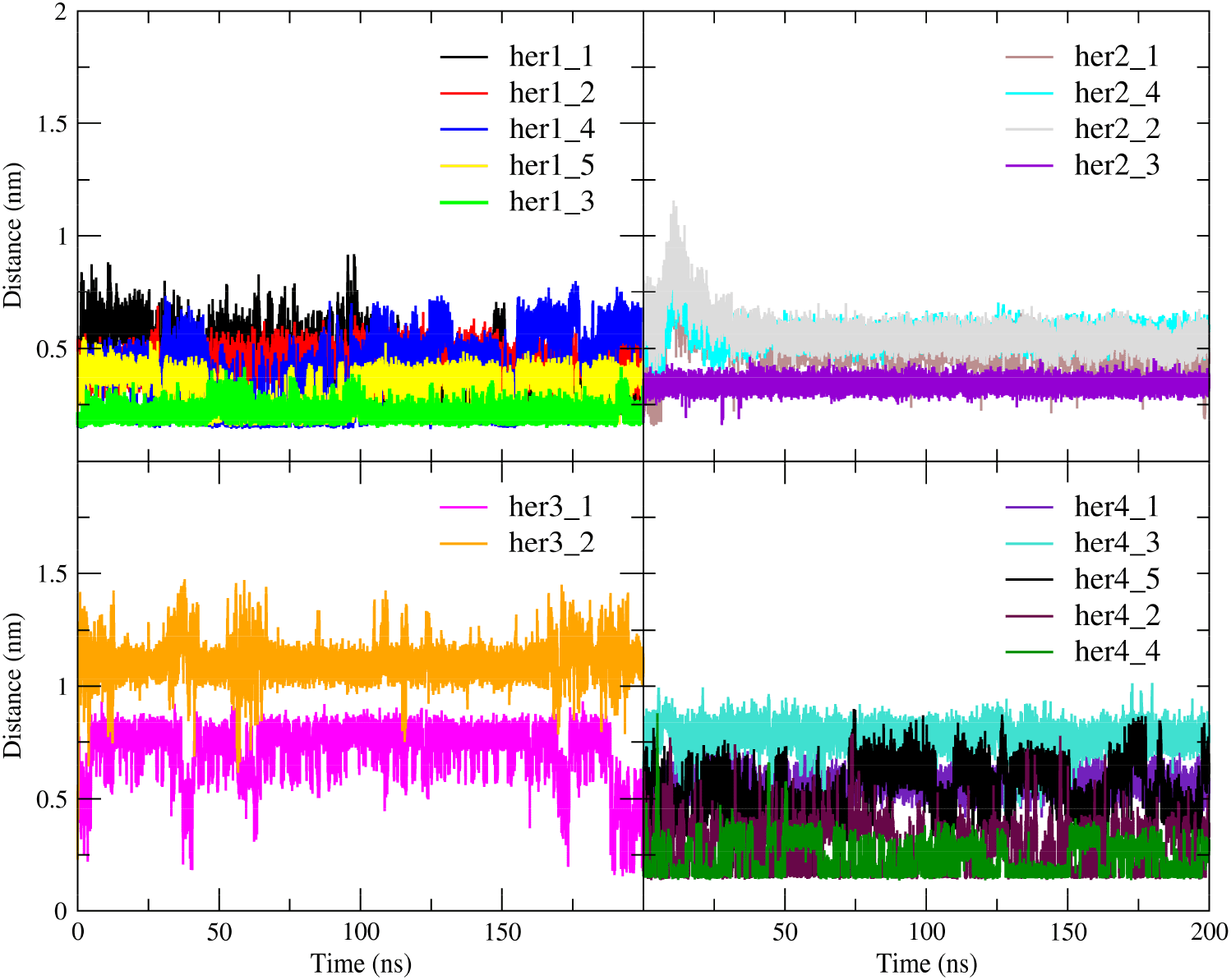
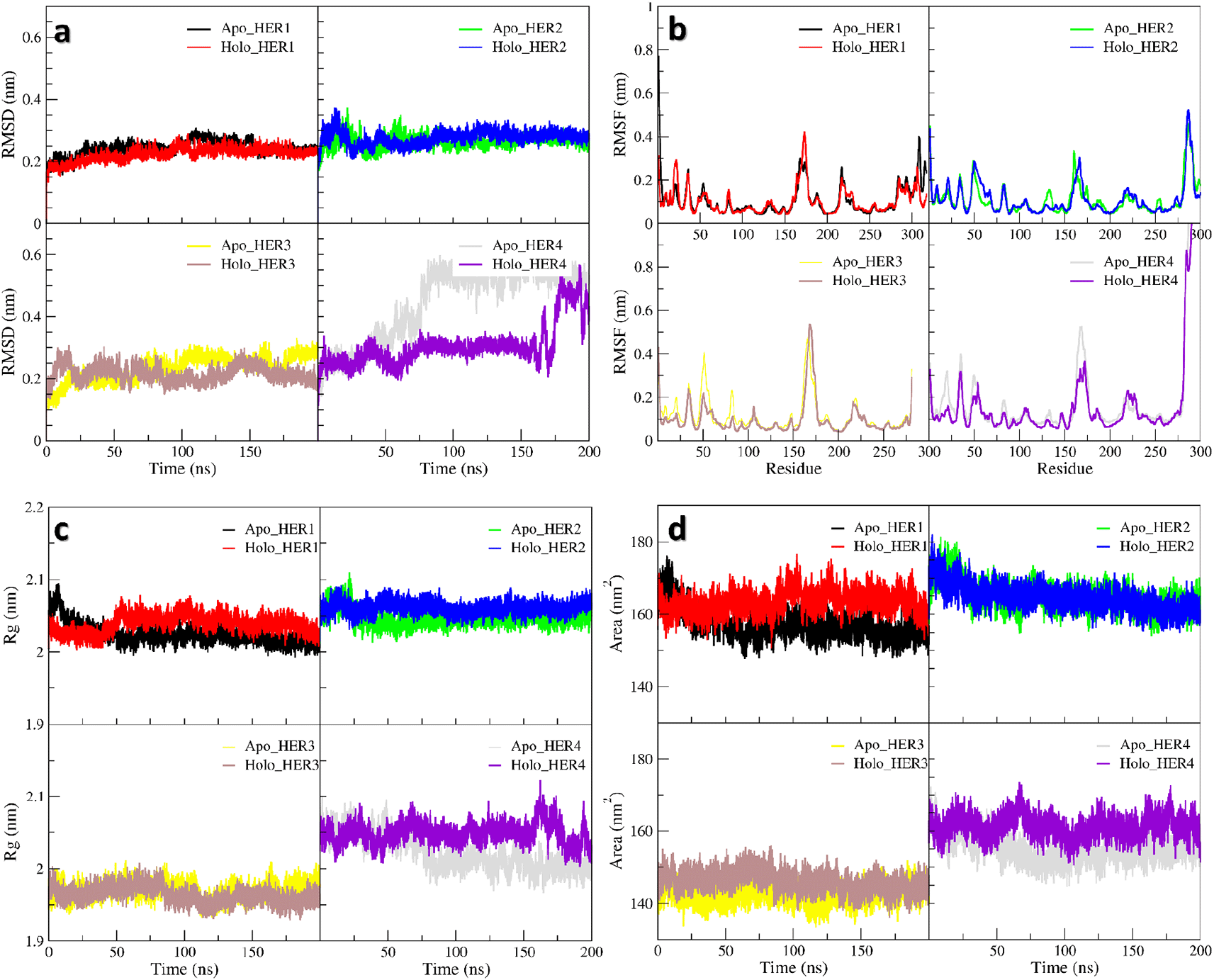
In all plots, the red and black lines represent holo-HER1 and apo-HER1, respectively. In all plots, the blue and green lines represent holo-HER2 and apo-HER2, respectively. In all plots, the brown and yellow lines represent holo-HER3 and apo-HER3, respectively. In all plots, the purple and grey lines represent holo-HER4 and apo-HER4, respectively.
PCA for the backbone of the receptors was performed and the first three eigenvectors were calculated and evaluated; the first two eigenvectors were taken for consideration as the most dynamical conformations present (Figure 6; Figure 8). Calculation and identification of the essential dynamic (PCA) is a method to monitor the motion of the protein to recognize its biological function. Also, PCA can help in making the trajectories shorter to spot the protein configurational spaces with irregular rhythm. To put it another way, PCA demonstrates, that a relatively small number of modes are sufficient to account for the majority of the fluctuations (Balsera et al., 1996). Fifty frames structures were isolated from each vector (one and two particularly) to facilitate spotting the change in the proteins structure (Figure 8). In terms of HER1, the PCA for the apo-protein showed that the region 164—180 contained the most observable irregular motion, and this region was duplicated in fluctuations caused by the binding of the ligand. Additionally, a portion of the 1—32 region was more pliable in the holo form (Figure 7). In contrast, the RMSF plot of vector one uncovered the fact that a portion of the region 199—232 was more flexible in its apo form than in its holo form. According to the results of the PCA plane analysis for the TKD of HER1, the apo form traveled through more configurational spaces than the holo form did. Whilst the PCA plane for the TKD of HER2 (Figure 6) postulated that both the holo and apo form visited different spaces at most, the significant difference in the dynamics was related to the region around residue 166, as revealed by the RMSF for the vector one, whereas the vector two had not shown any change in its behavior. Mulberroside A did not exert a significant influence on TKD of HER3 due to the fact that the principal plane demonstrated a stability and occupation almost for the same configurational spaces regarding holo and apo-form (Figure 6). As can be seen in the RMSFs of both vector one and vector two, the small change in the conformation that occurred as a result of the ligand's occupancy was primarily attributable to a reduction in the flexibility of the multiple parts that make up region 1—100. According to the principal plane of the apo form, which completely visited different spaces with a very wide transition in the conformation in comparison to the holo-form, TKD of HER4 dramatically changed its conformations upon interaction with the ligand. The change was primarily represented by the C-terminal and the portion between 160 and 180. When these receptors are activated, their C-terminal regions undergo autophosphorylation and act as a binding site for signaling proteins that transfer the signal downstream (Kovacs et al., 2015; Monsey et al., 2010).
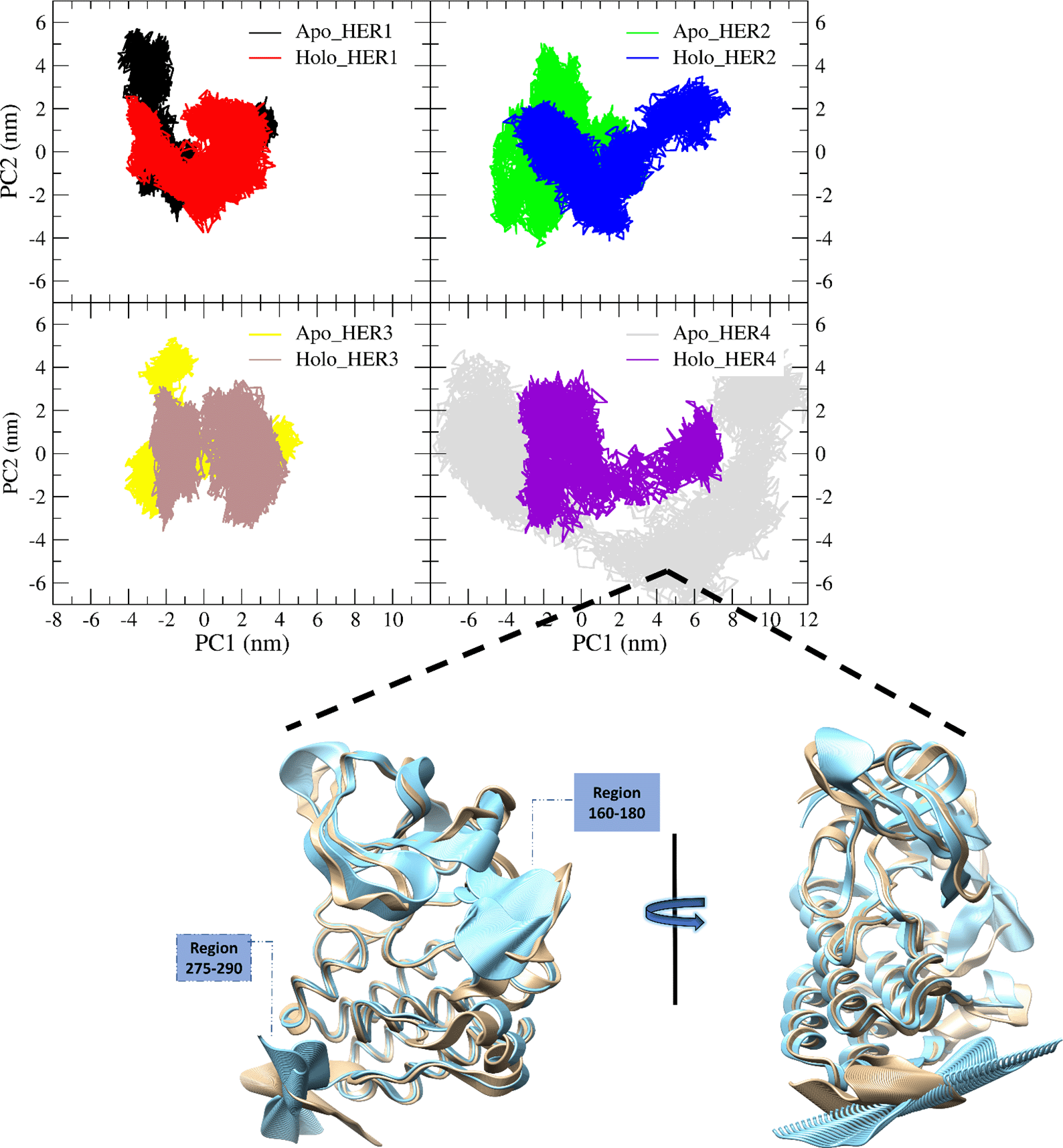
Concerning HER4, the fifty frames of vector one for both the apo (blue) and holo (khaki) forms were matched to better visualize the major conformational difference between them. These changes appear to originate from the highly fluctuated region 160—180 and the C-terminus.
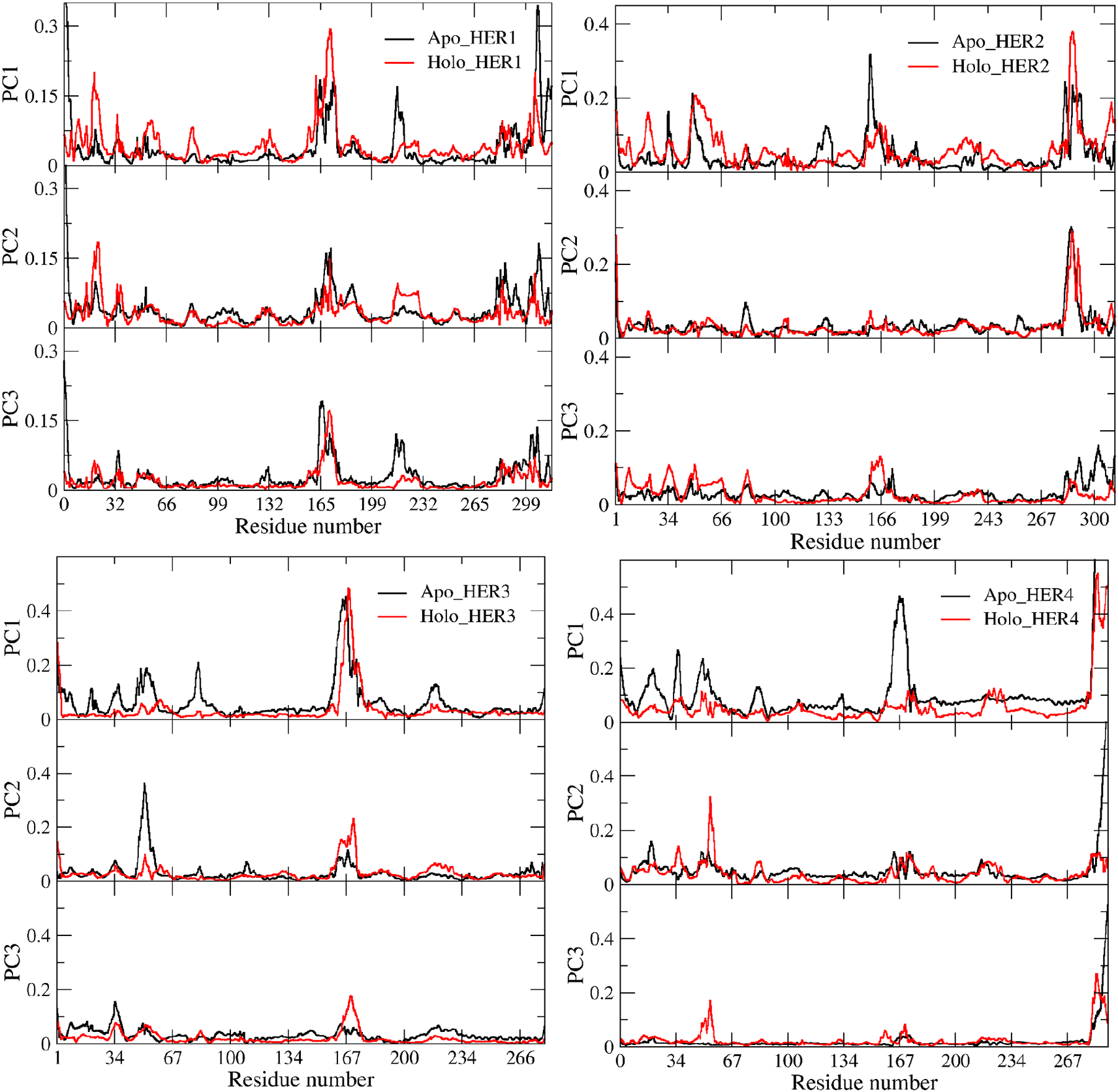
The holo proteins are shown in red, while the apo proteins are shown in black. The docked and undocked HER1 is shown in the upper left square, the docked and undocked HER2 is shown in the upper right square, the docked and undocked HER3 is shown in the lower right square, and the docked and undocked HER4 is shown in the lower left square.
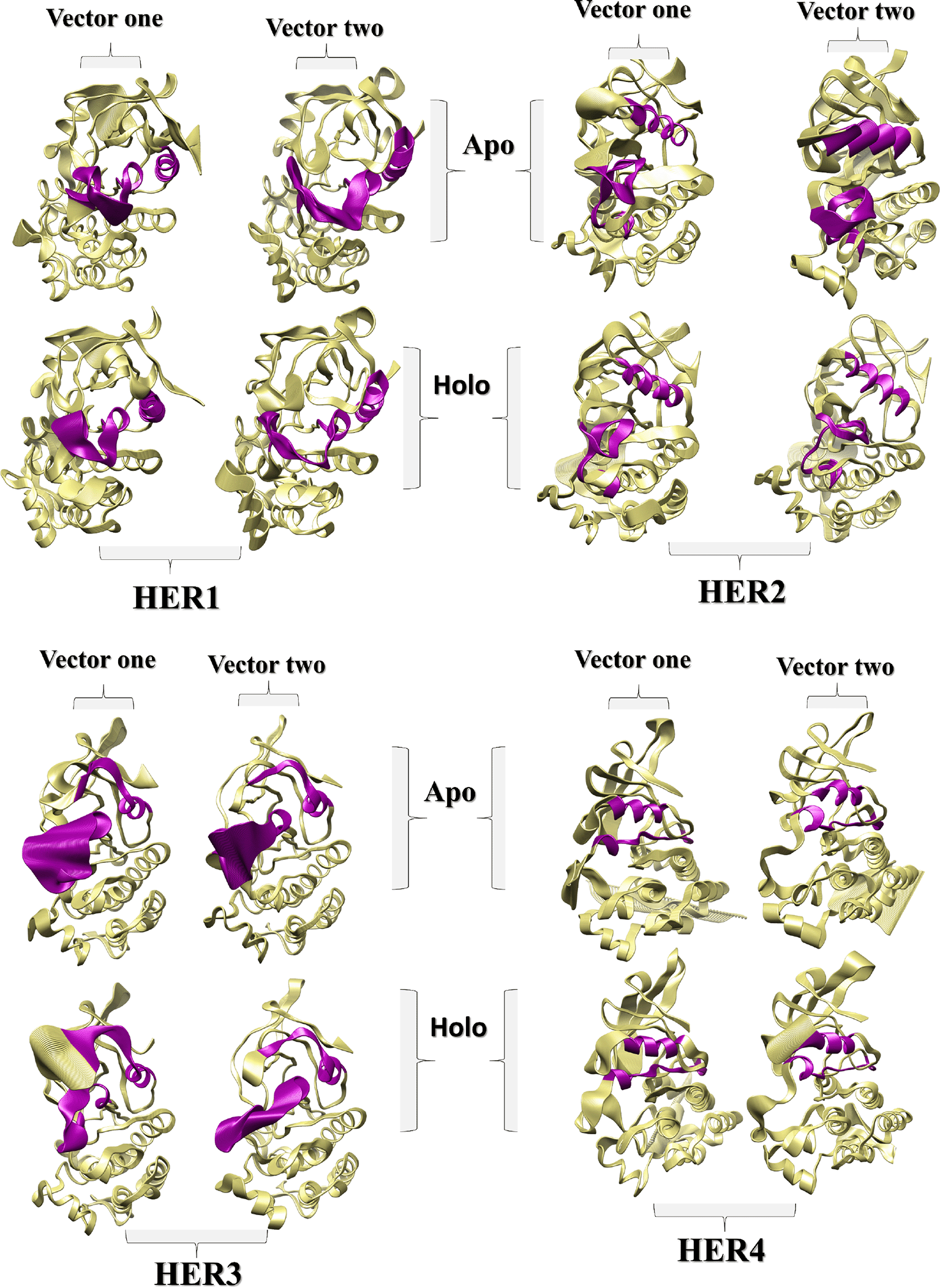
The C -helix and the activation loop are colored purple, as should be noted. As illustrated by the aligned frames, the degree and pattern of the activation loop's fluctuation differ slightly between the holo and apo forms of HER1, as does the orientation of the C α-helix. Upon the binding of the ligand to HER2, the decrease in HER2's flexibility is readily apparent. The flexibility of HER3's activation loop and neighboring regions appears to be greater than that of other proteins, both in the occupied and unoccupied state. Regarding HER4, the degree and pattern of fluctuation for the C -helix and the activation loop did not change significantly, with the exception of the regions shown in Figure 6.
The dearth of chemotherapeutic agents that are still effective against cancers through the tyrosine kinase receptors due to pervasive resistance is a significant issue for the scientific community. Mulberroside A a natural polyphenolic compound has been approved for its capability to bind tightly to these receptors leading to significant changes in the dynamic of the vital parts of their structures. RMSD, RMSF, Rg, and SASA calculations have revealed a change in the conformation, compactness, and the folding pattern of the TKDs upon the binding of mulberroside A. These findings were also supported by the PCA, which elucidated a variation in the dynamic of the backbone of the proteins. Due to the polar properties of the binding site of the human epidermal receptors, the polyphenolic compound mulberroside A perfectly interacted with them. The C α-helix changed in behavior upon the binding of the ligand to the TKD in HER1, HER2, and HER3, which may lead to a change in the catalytic activity of the enzyme. The activation loop is affected as well in HER1, HER2, and HER3 and could lead to a prediction of changing the activation level of these enzymes. Regarding HER4, the region that was affected by the ligand is different and may affect the autophosphorylation process. Broadly speaking, mulberroside A is a promising tyrosine kinase domain inhibitor, and the HERs overexpressed cell-based assay is recommended for further investigation of its in-vitro activity.
Protein sequences and structures are taken from Protein Data Bank:
- Accession number: Protein Data Bank, PD3POZ
- Root URL: https://identifiers.org/pdb
- Accession number URL: https://identifiers.org/pdb:3POZ
- Accession number: Protein Data Bank, PD3RCD
- Root URL: https://identifiers.org/pdb
- Accession number URL: https://identifiers.org/pdb:3RCD
- Accession number: Protein Data Bank, PD6OP9
- Root URL: https://identifiers.org/pdb
- Accession number URL: https://identifiers.org/pdb:6OP9
- Accession number: Protein Data Bank, PD3BBT
- Root URL: https://identifiers.org/pdb
- Accession number URL: https://identifiers.org/pdb:3BBT
The following link contains all molecular dynamic parameter (mdp) files:
https://drive.google.com/drive/folders/1zdkRAnzupDKNjmojd7n-vR3EkdZiq5Rd?usp=sharing
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)