Keywords
Veterinary SWATH analysis, peptide spectral library, sheep serum proteomics, TICs, annotation of proteins in serum, nanoLC-nanoESI-MS/MS, sick sheep, healthy sheep
Veterinary SWATH analysis, peptide spectral library, sheep serum proteomics, TICs, annotation of proteins in serum, nanoLC-nanoESI-MS/MS, sick sheep, healthy sheep
ACN: Acetonitrile
ALP: Alkaline phosphatase
AST: Aspartate aminotransferase
ATP: Adenosine triphosphate
AUC: Area under curve
BCA: Bicinchoninic acid assay
CARF: Central Analytical Facility
CCKR: Cholecystokinin receptors
CK: Creatine Kinase
DIA: Data independent acquisition
DTT: Dithiothreitol
EBI: European Bioinformatics Institute
EGF: Epidermal growth factor
EMBL: European Molecular Biology Laboratory
FA: Formic acid
FGF: Fibroblast growth factor
GGT: Gamma-glutamyl transferase
GO: Gene ontology
IAM: Iodoacetamide
ID: Identification
LC: Liquid chromatography
MS/MS: tandem mass spectrometry
MS: Mass spectrometry
nanoLC-nanoESI-MS/MS: nano liquid chromatography nano electrospray ionisation tandem mass spectrometry
PANTHER: Protein ANalysis THrough Evolutionary Relationships
PCA: Principal component analysis
PRIDE: Proteomics identifications
PSL: Peptide spectral library
QUT: Queensland University of Technology
RT: Room temperature
SWATH: Sequential acquisition of all theoretical fragrant mass spectra
TEMED: Tetramethylethylenediamine
TFA: Trifluoroacetic acid
TOF: Time-of-flight
TP: Total protein
UQ: The University of Queensland
Until now, the use of sequential window acquisition of all theoretical fragment mass spectra (SWATH-MS) analysis1 to complement traditional shotgun MS-based proteomics2 on samples from species of veterinary importance has been uncommon. Only a limited number of published reports have applied this approach on animal experimental subjects, for example in the quantification of hepatic proteins of chicken exposed to heat stress,3 analysis of seminal plasma protein of pigs,4 identification of proteins involved in nutritional stress in goats,5 quantification of protein alterations in plasma of acutely endotoxaemic sheep,6 and detecting proteins in peptide spectral libraries of bovids.7 Otherwise, much of the work in this area has been centred upon in-vitro studies on cell lysates8–11 and body fluids obtained from mice.12 Despite its promising nature in protein analysis, SWATH-MS analysis remains largely untested on actual clinical samples from veterinary patients.
In this report, SWATH-MS was used to analyse serum samples obtained from sheep suffering from a range of naturally-acquired illnesses and compared with samples from healthy sheep, by interrogating a novel peptide spectral library (PSL) constructed from the ovine circulating acellular proteome.6,13 The sick sheep serum samples were of actual clinical cases submitted to The University of Queensland (UQ) (Gatton, Australia). Serum samples from twenty healthy sheep were obtained from a commercial source (Serum Australis Pty Ltd.) and normalised to represent an analytical normal proteome background. Tryptic peptides were purified by StageTip technique,14,15 prior to nano-liquid chromatography nano-electrospray ionisation tandem mass spectrometry (nanoLC-nanoESI-MS/MS) in a cyclic data-independent acquisition (DIA) mode, using a generic SWATH-MS acquisition method (SWATH™, SCIEX). The acquired data were processed in PeakView® software with SWATH™ Acquisition MicroApp 2.0 (SCIEX) pipeline to generate protein lists for downstream gene ontology annotation and pathway analysis of identified proteins.
The use of SWATH-MS analysis enabled protein profiles of individual sick sheep to be differentiated as compared to the normalised healthy sheep serum aliquots. This approach detected some established clinical biochemical analytes and possibly other candidate disease markers. These observations suggest that it is potentially feasible to use SWATH-MS in conjunction with the novel PSL to determine relative protein alterations in clinical samples from veterinary patients in future.
The experiments included in this study were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.16 Animal tissue samples for the generation of the PSL were acquired following approval from Queensland University of Technology (QUT) Confirmation number: 1400000591, QV reference number: 46375 and Ethics number TRIM 10/8428 issued to Serum Australis Pty Ltd (http://www.serumaustralis.com.au), for bleeding live sheep and supply of blood products. Tissue from live animal experiments had animal ethics approval obtained from the University Animal Ethics Committee of QUT (Reference No. 0800000555), which was ratified by The University of Queensland (UQ).
Ex-diagnostic serum samples of seven sheep that presented with a range of clinical manifestations each, were opportunistically obtained from UQ laboratory archive for analysis. The accompanying clinical details of these sheep are presented in Additional file 1.48 In this set of experiments, the individual code-identified samples from each sheep (SCi502, SCi503, SCi504, SCi507, SCi508, SCi509 and SCi510), a pooled sample of all of them (SCi511) and a pooled sample of aliquots from 20 healthy sheep obtained from Serum Australis Pty Ltd (SA) (SCi512) as the normalised analytical control for the experiments, were prepared and subjected to proteomic analysis using the SWATH-MS (SCIEX) analysis pipeline (Figure 1).
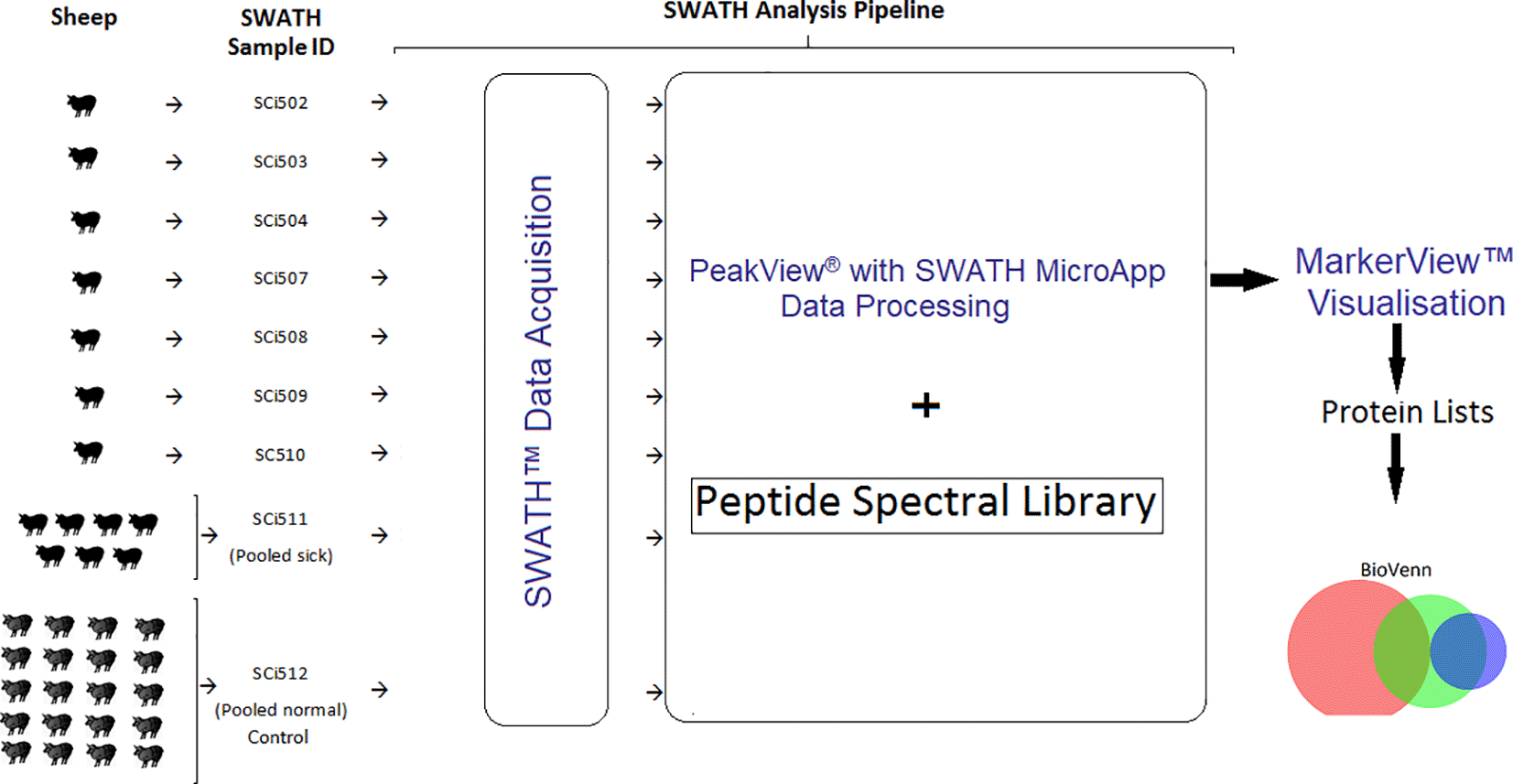
Serum samples derived from seven sick sheep (SWATH Sample ID: SCi502, SCi503, SCi504, SCi507, SCi508, SCi509 and SCi510), a pooled sample from all the sick sheep (pooled sick) (SWATH Sample ID SCi511) and a normalised sample pooled from 20 healthy sheep (pooled normal) (SWATH Sample ID SCi512). The acquired SWATH data from each sample were processed in PeakView® v2.2 software with SWATH™ Acquisition MicroApp 2.0 (SCIEX) – SWATH pipeline. Data were exported into MarkerView™ v1.3 software (SCIEX) for statistical analysis protein list generation. Protein lists were exported to spreadsheet (Microsoft® Excel™), which facilitated comparative analysis using a Venn diagram (BioVenn software).
Samples were prepared as in a previously described method.13 In brief, frozen sheep serum samples were thawed and kept under refrigeration conditions with an added protease inhibitor (cOmplete™, Roche) according to the manufacturer’s instructions. Each sample was agitated vigorously for half a minute before centrifuging at 13,000 g for 20 minutes. Only the supernatant was retained for downstream analysis, after determining the protein concentration with bicinchoninic acid (BCA) protein assay method (BCA Protein Assay Kit, Pierce™) and a spectrophotometer (NanoDrop 2000; Thermo Scientific).
Proteins in the sample supernatants were precipitated by adding 4 × (v:v %) of cold acetone (20 °C) and then incubated at this temperature for 16 h, followed by centrifugation at 4,000 g for 2 minutes. The pellet was then retained and washed using 1 mL of cold acetone by agitating vigorously to break the pellet. The suspension was then centrifuged at 4,000 g for 5 min at 4 °C and sediment was retained. This washing procedure was performed once more. The pellet was dissolved in aqueous fresh 8 M urea in 25 mM ammonium bicarbonate (NH4HCO3) and then centrifuged at 4,000 g for 5 min at 4 °C. The protein concentration of the supernatant was then determined by BCA protein assay.
While still under refrigeration conditions, sample aliquots comprising 100 μg of protein were reduced with 20 mM DTT (titrated to 5 mM final concentration) and then incubated for 1h at room temperature (RT), followed by alkylation with 55 mM iodoacetamide (IAM) (resulting in 14 mM final concentration) and incubated again for 20 min in the dark at RT. Alkylation was suppressed using dithiothreitol (DTT) prior to further incubation for 5 min in the dark after which the solution was diluted with 25mM NH4HCO3 buffer followed by titrating aqueous 70 mM CaCl2 into the samples (aiming at 10 mM final concentration). Trypsin (Promega) was added at a ratio of 1:50 (enzyme to protein concentration) and the contents were incubated for 16 h at 37 °C with gentle shaking and then cooled to RT. Protein digestion was curtailed using 10% trifluoroacetic acid (TFA) before vacuum drying the contents. Peptides were then dissolved in aqueous 0.1% TFA/2% acetone nitrile (ACN) and routinely desalted using octadecyl carbon chain (C18) pipette tips (ZipTip® Pipette Tips, Millipore), vacuum dried, reconstituted in 10 uL of aqueous 2% ACN/0.1% FA for nanoLC-nanoESI-MS/MS analysis.
Quantities of about 400 ng – 1 μg of peptides per sample were subjected to reversed-phase chromatography setup in a trap and elute arrangement across a 90 min gradient at 40 °C. Two mobile phases A and B were used, prepared from aqueous 0.1% formic acid (FA) and ACN/0.1% FA, respectively on a nanoLC-nanoESI-MS/MS system (TripleTOF® 5600+ instrument (SCIEX)).
Spectral data of purified eluted peptides were extracted by cyclic DIA using a generic SWATH™ acquisition method described elsewhere.1,3 The mass spectrometer was operated using 0.1 s for the survey MS run, followed by tandem mass spectrometry on all precursors in a recurring manner with an accumulation time of 0.1 s per SWATH window across 36 windowsl, each 26 m/z units wide for total cycle duration of 3.75 s in order to sample at least 6 data points for each chromatographic peak per peptide to ensure improved quantification precision.
The proprietary raw data files (.wiff format) were concurrently imported into PeakView® v.2.2 software with integrated SWATH™ Acquisition MicroApp 2.0.1 (SCIEX) for spectral alignment and targeted data extraction from the PSL to be interrogated. The PSL was assembled from hundreds of samples derived from serum and plasma of sick and healthy sheep that had hitherto been analysed by nanoLC-nanoESI-MS/MS by data dependent acquisition (DDA) on the same instrument as described in earlier.6,13 Extracted ion chromatograms (XICs) from MS/MS spectra for targeted peptides were generated by the SWATH™ Acquisition MicroApp 2.0.1, by assimilating peak areas from the SWATH™ data files as detailed in the vendor’s technical note (http://tinyurl.com/j6w83pu).
The parameters for SWATH™ data extraction were set as follows: five peptides per protein, five transitions per peptide, peptide confidence level of >95%, exclude shared peptides, and extracted ion chromatograph (XIC) width of 50 ppm. Processed data were exported as quantitative output for the peak area under the intensity curve for individual ions, the summed intensity of individual ions for a given peptide, and the summed intensity of peptides for a given protein in .txt format. Data were then statistically analysed using MarkerView™ software (SCIEX). Data were then exported in.xlxs format (Microsoft® Excel™) into spreadsheet for inspection and comparative visual analysis using BioVenn software.17
There are other open-access alternative software that can be used to for analysis of the data used in this article, for example the Trans-Proteomic Pipeline (TPP),18 Skyline19 and OpenSWATH.20,21
The total ion chromatograms of each of the analysed peptides derived from serum samples of sick sheep were processed in PeakView® software and compared with that of healthy sheep (Additional file 248). A total of 335 proteins (Additional file 3(a)48) were identified in the healthy sheep sample pool (SCi512) that was derived from aliquots of 20 sheep, representing the normal proteome background. Protein identifications (IDs) from individual samples of sick sheep were compared to the IDs of SCi512 to reveal shared and unshared overlaps in protein IDs, and total combined protein IDs in each individual clinical case experiment as shown in Figure 2A. A comparison of protein IDs from pooled normal sheep serum (SCi512), pooled sick sheep (SCi511) and those unique to sick sheep only is presented Figure 2B. There were proteins that were unique to each of the sick sheep samples, as compared to healthy sheep pool. These unique proteins numbered 55, 95, 61, 54, 80, 61, 30, and 70 for SCi502, SCi503, SCi504, SCi507, SCi508, SCi509, SCi510, and SCi511, respectively. The UniProtKB entries for the individual proteins unique to each sick sheep sample are presented in Additional file 3 (b), (c), (d), (e), (f), (g), (h) and (i)48, respectively.
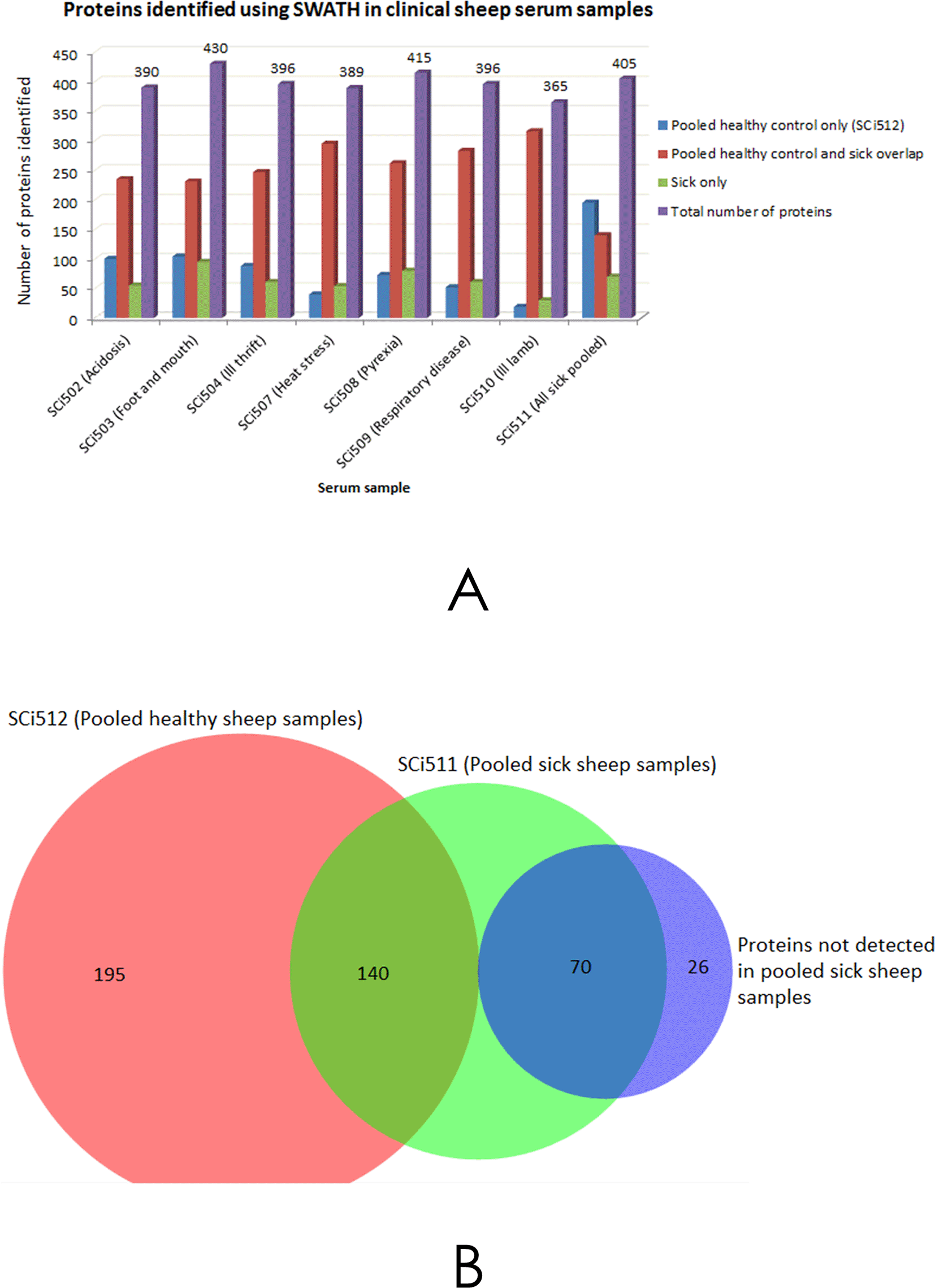
A - Shows the number of protein IDs obtained using SWATH™ processing of individual serum samples SCi504, SCi503, SCi504, SCi507, SCi508, SCi509 and SCi510 from sick sheep, and a pooled sample from all the preceding samples (SCi511), that were compared with a pooled serum sample from healthy sheep (SCi512). B - Illustrates a composite analysis of all the samples that yielded 431 protein IDs, 335 of these IDs were attributed to SCi512, 210 IDs were from the pooled sick sheep sample (SCi511) and 96 IDs were exclusive to sick sheep serum only. The 26 protein IDs that were not detected in SCi511 were identified by analysing proteins that were exclusive to sick sheep (sick sheep only fractions) of the individual samples in A.
The UniProtKB entries for the 195 proteins that were identified as being unique to the healthy sheep sample pool (i.e. proteins that were not detected in sick sheep samples illustrated in Figure 2B), are listed in Additional file 3 (j).48 Also listed, are the 96 protein IDs (Additional file 3 k48) that were unique to sick sheep and the 26 protein IDs (Additional file 3 (l)48) that were not detected in the sick sheep pool (SCi511) compiled from protein IDs exclusive to the sick sheep samples. The UniProtKB entries for all of the 431 proteins identified in the entire workflow comprising of serum samples from sick and healthy sheep in Figure 2B are listed in Additional file 3 (m).48
There was differential abundance in proteins identified across all the analysed samples, including protein IDs from healthy sheep. The relative intensities of the top 10 most abundant protein IDs in healthy sheep serum pool in descending order, alongside protein IDs from the seven individual sick sheep samples (SCi502, SCi503, SCi504, SCi507, SCi508, SCi509 and SCi510), and the pooled sample from sick sheep (SCi511) are illustrated in Figure 3. The relative abundance of many proteins was variable between the different samples as presented in Additional file 4.48 There were notable differences in protein intensities between the healthy sheep serum pool and the sick sheep sample pool as shown in Figure 4.
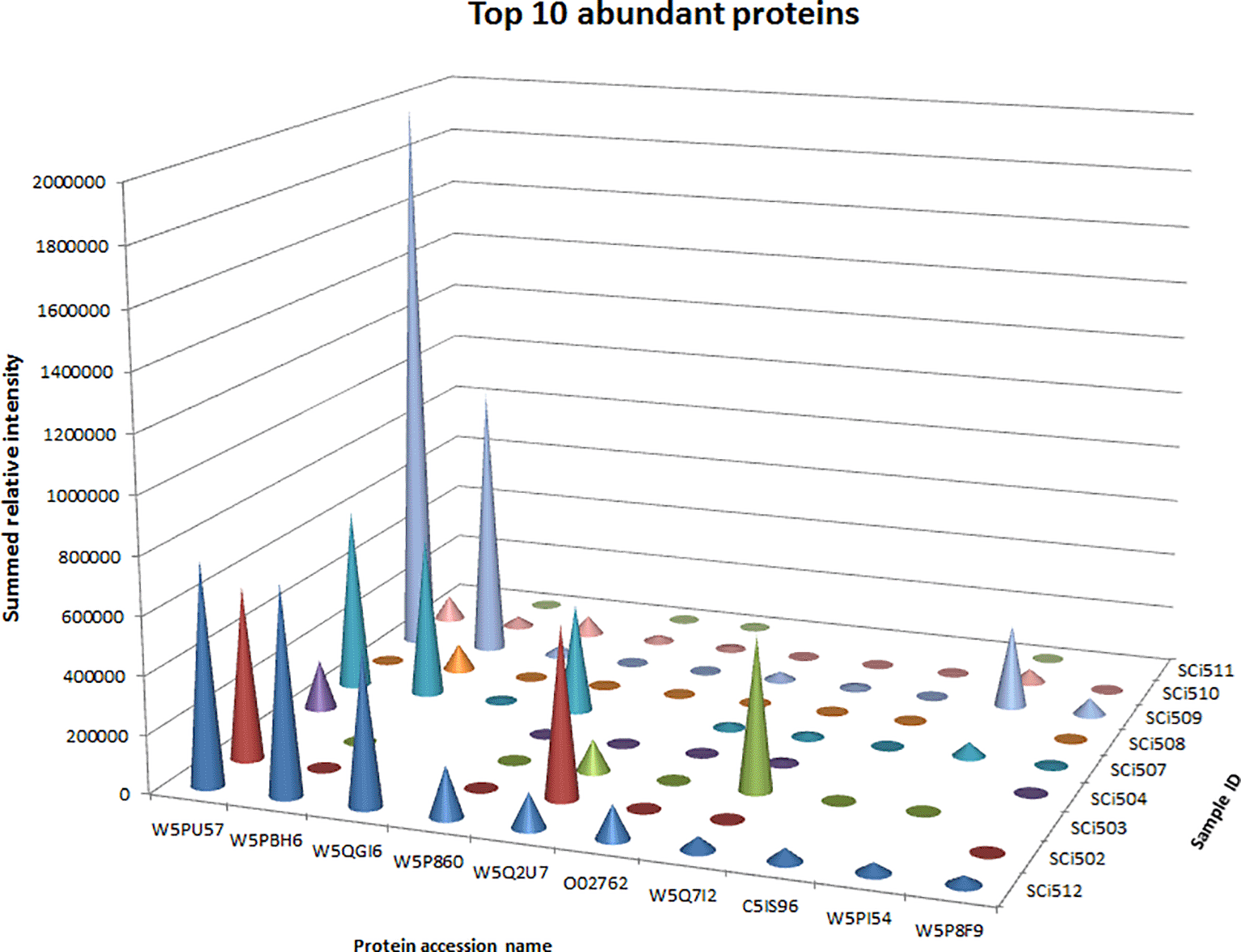
Protein intensities from a pooled sample of healthy sheep (SCi512) in descending order were compared with intensities from sick sheep samples (SCi502, SCi503, SCi504, SCi507, SCi508, SCi509, SCi510 and SCi511 (pooled sick)) and corresponding proteins.
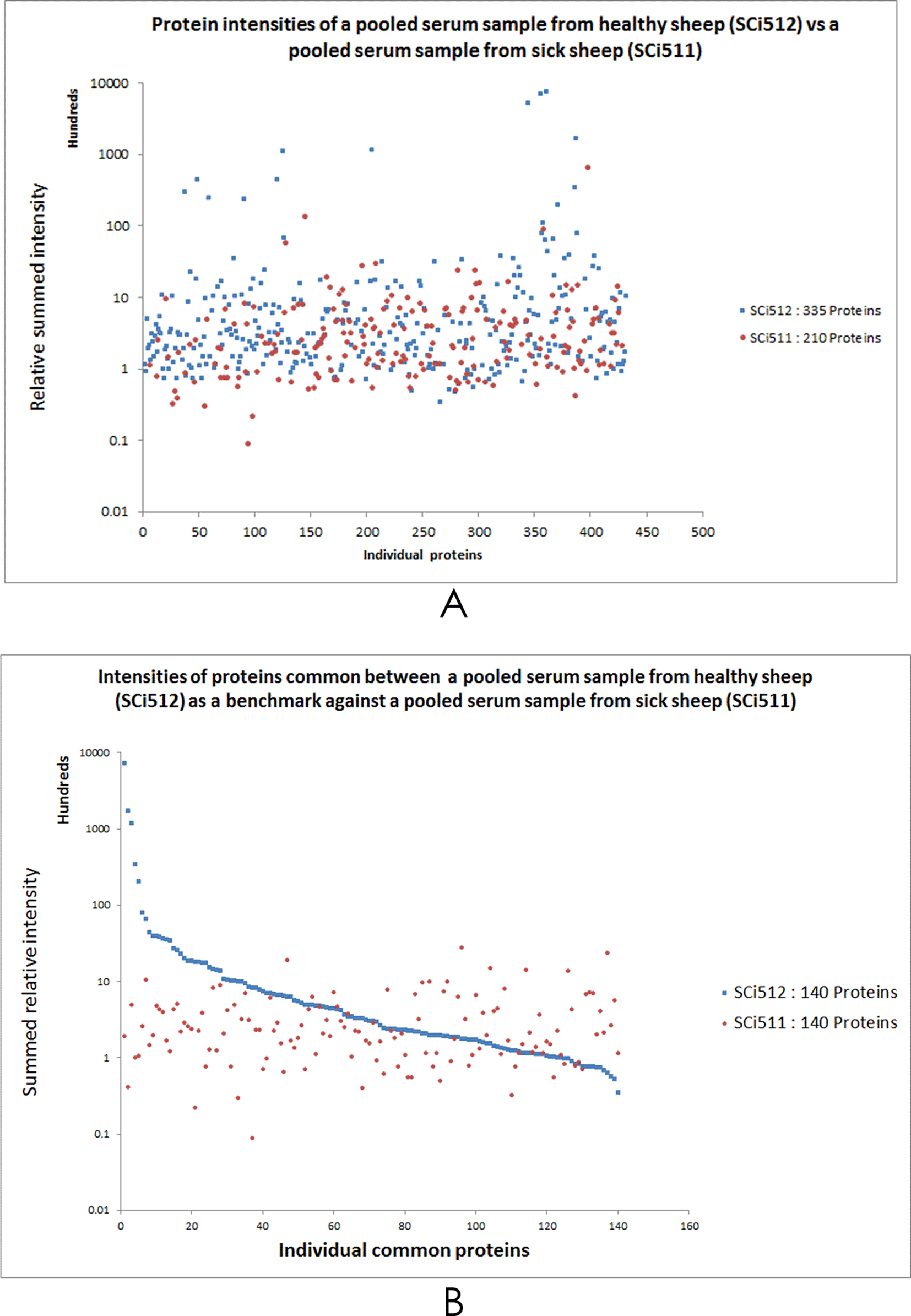
A – Shows the relative summed protein intensities of all proteins identified by SWATH-MS analysis in a pooled sample from healthy sheep (SCi512) as compared with a pooled sample (SCi511) derived from 7 samples (SCi502, SCi503, SCi504, SCi507, SCi508, SCi509 and SCi510) from sick sheep. B – Shows the quantitative comparison between the two samples representing healthy sheep and sick sheep was possible, however only 140 identified proteins were common between SCi512 and SCi511. In (B), taking the protein intensities calibration plot from SCi512 as the normalised control (or benchmark), protein IDs in sample SCi511 falling above the blue line were considered upregulated, whilst those below this line were designated as downregulated. Note the wide dynamic range in the intensities of some proteins in both (A) and (B).
Of the 335 proteins identified in the healthy sheep serum pool, the ten most abundant proteins (see Figure 2B and Additional file 3(a)48) in descending order were W5PU57 (Nuclear envelope pore membrane protein POM 121C), W5PBH6 (RAD54 like 2), W5QGI6 (Exonuclease 3'-5' domain containing 1), W5P860 (Dual-specificity kinase), W5Q2U7 (Plectin), O02762 (Apolipoprotein A1), W5Q7I2 (I Ig-like domain-containing protein), C5IS96 (Lecithin-cholesterol acyltransferase), W5PI54 (Rabphilin 3A) and W5P8F9 (BPI1 domain-containing protein). The ten least abundant proteins in this healthy sheep sample pool were W5P0B2 (LDL receptor related protein 2), W5QBV7 (CD44 antigen), W5PAS6 (EvC ciliary complex subunit 2), W5QCV8 (Cadherin EGF LAG seven-pass G-type receptor 2), W5P640 (Lamin A/C), W5PQ96 (Ciliary rootlet coiled-coil, rootletin), W5P7B1 (Sirtuin 2), W5QGV5 (Dedicator of cytokinesis 10), W5QGX2 (Spectrin beta, non-erythrocytic 5) and A2P2G4 (VH region).
Of the 210 proteins identified in the sick sheep serum sample pool (SCi511)(Figure 2B), the ten most abundant proteins in descending order were W5QIV1 (Protein S100), W5QHZ8 (Ig-like domain-containing protein), W5PGA9 (Nicastrin), W5PI92 (Apolipoprotein C-IV), W5NV45 (Hephaestin like 1), A2P2I4 (VH region), W5P640 (Lamin A/C), B0FZL9 (Pre-mRNA splicing factor SRP20-like protein), W5PVM3 (Myocilin) and W5NQ83 (RNA binding motif protein 25). The ten least abundant proteins in this sample were W5PJG0 (Serum amyloid A protein), W5NX96 (Attractin), W5PV45 (Centrosomal protein of 162 kDa), W5PBY0 (Complement component 4 binding protein alpha), W5P860 (Dual-specificity kinase), P12303 (Transthyretin), W5QFP0 (Thrombospondin 1), W5P3J3 (Complement C1s), W5NXP3 (Serpin A3-6-like) and W5PHP8 (Leucine rich alpha-2-glycoprotein 1).
All the 236 proteins identified from a composite of all sick sheep serum samples and the 335 proteins from healthy sheep pool (Figure 2B) — taking into consideration overlapping protein IDs, were subjected to gene ontology (GO) classification, within the domain terms of molecular function, biological process, cellular component, protein class using the PANTHER (Protein ANalysis THrough Evolutionary Relationships) classifications system24 (Figure 5). Using the UniProtKB gene identification and mapping tool, 215 of the 236 proteins identified in sick sheep serum were mapped to 206 named gene IDs of sheep. Of these identified sheep genes, only 172 of them were recognised by the PANTHER tool after aligning them to Bos taurus — the species with a genome most closely homologous that of sheep. Likewise, 299 of the 335 proteins identified in healthy sheep serum pool were mapped to 290 sheep genes, however, only 251 genes were recognised in PANTHER based on bovine homologous entries.
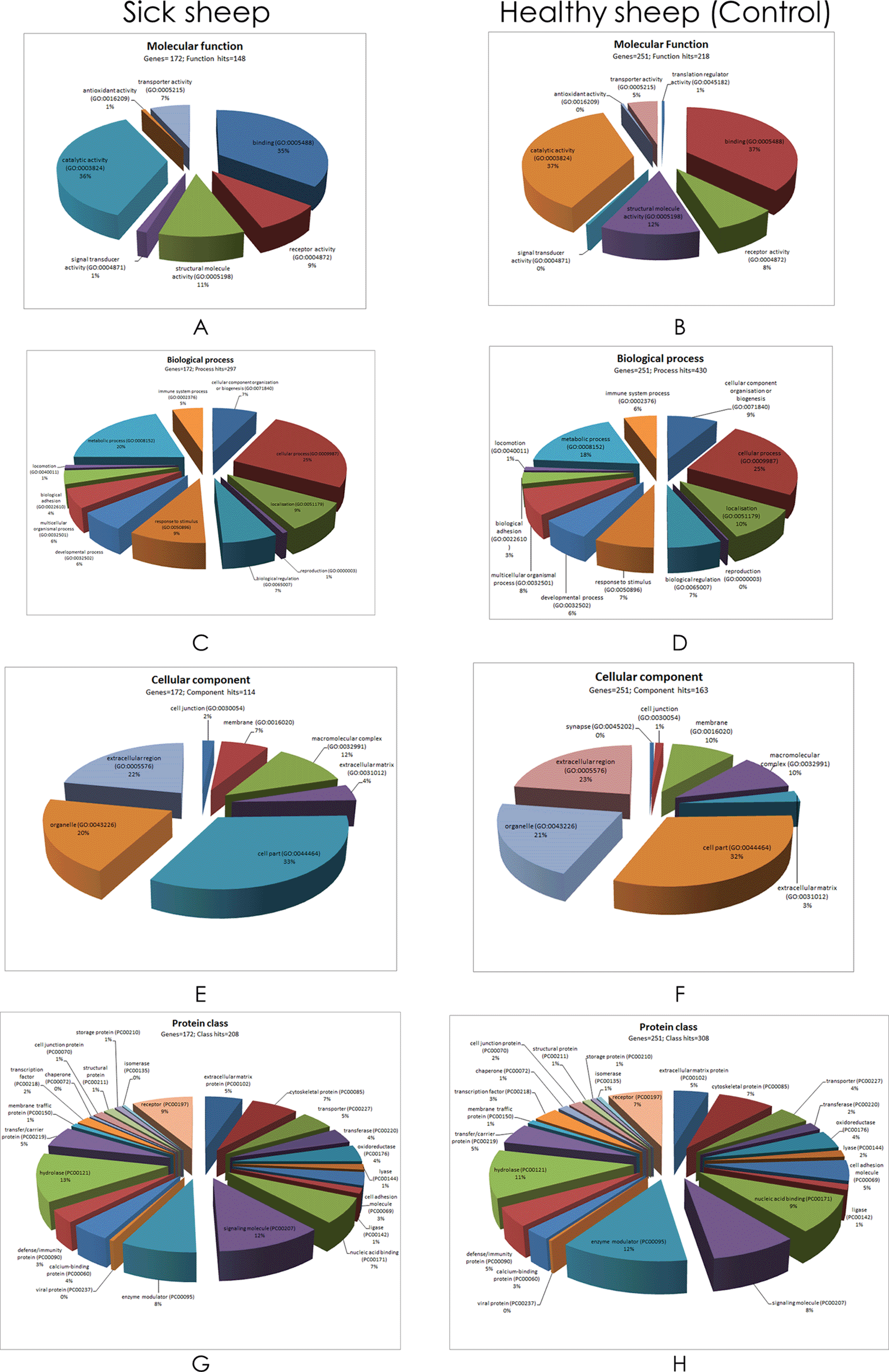
Note that the number of genes and individual GO term hits were different between sick and healthy sheep pool.
Apart from the number of distinct genes and individual GO term hits, the fractional representation (percentages) of the contributing elements within the domains between sick and healthy sheep serum were comparable (Figure 5). Catalytic activity, binding, structural molecule and receptor activity had comparatively larger representations in the molecular function domain. Cellular process, metabolic process, response to stimulus and localisation had the largest representation in the biological process domain. In the cellular component domain, cell part, extracellular region, organelle and macromolecular complex had the largest representation. As for the protein class domain, enzyme modulator, hydrolase, signalling molecule and receptor terms had the largest representation of the GO terms. It was evident from the molecular function domain that large representations of proteins are involved in catalytic activity (36%) and binding (35%). The biological process domain is interesting for these case studies that involve pathology in that it illustrates inclusion of immune process (5%) and response to stimulus (9%), alongside metabolic (20%) and cellular processes (25%). Cell part (33%), extracellular region (22%) and organelle (20%) GO terms were predominant in the cellular component domain. Meanwhile defense/immunity comprised of only 3%, compared to hydrolase (13%) signalling molecule (12%), receptor (9%) and oxidoreductase (4%) terms in the protein class domain.
Protein pathway analysis was conducted on all the identified proteins from healthy sheep serum sample pool and sick sheep serum using PANTHER (Figure 6). Differences were observed in the enriched protein pathways between sick and healthy sheep. In the results from serum derived from sick sheep, the predominantly represented protein pathways were ATP synthesis, interleukin signalling, angiogenesis, Alzheimer disease-presenilin, integrin signalling, inflammation mediated by chemokine and cytokine signalling, gonadotropin-releasing hormone receptor, cytoskeletal regulation by Rho GTPase, blood coagulation, Huntington disease, p53 pathway, Wnt signalling pathway and Glycolysis pathways. As for the serum derived from healthy sheep, the predominantly represented protein pathways were Alzheimer disease-presenilin pathway, integrin signalling, inflammation mediated by chemokine and cytokine signalling pathway, Huntington disease, Wnt signalling pathway, glycolysis, Parkinson disease, cytoskeletal regulation by Rho GTPase, cadherin signalling, blood coagulation, cholecystokinin receptors (CCKR) signalling map and gonadotropin-releasing hormone receptor pathways.
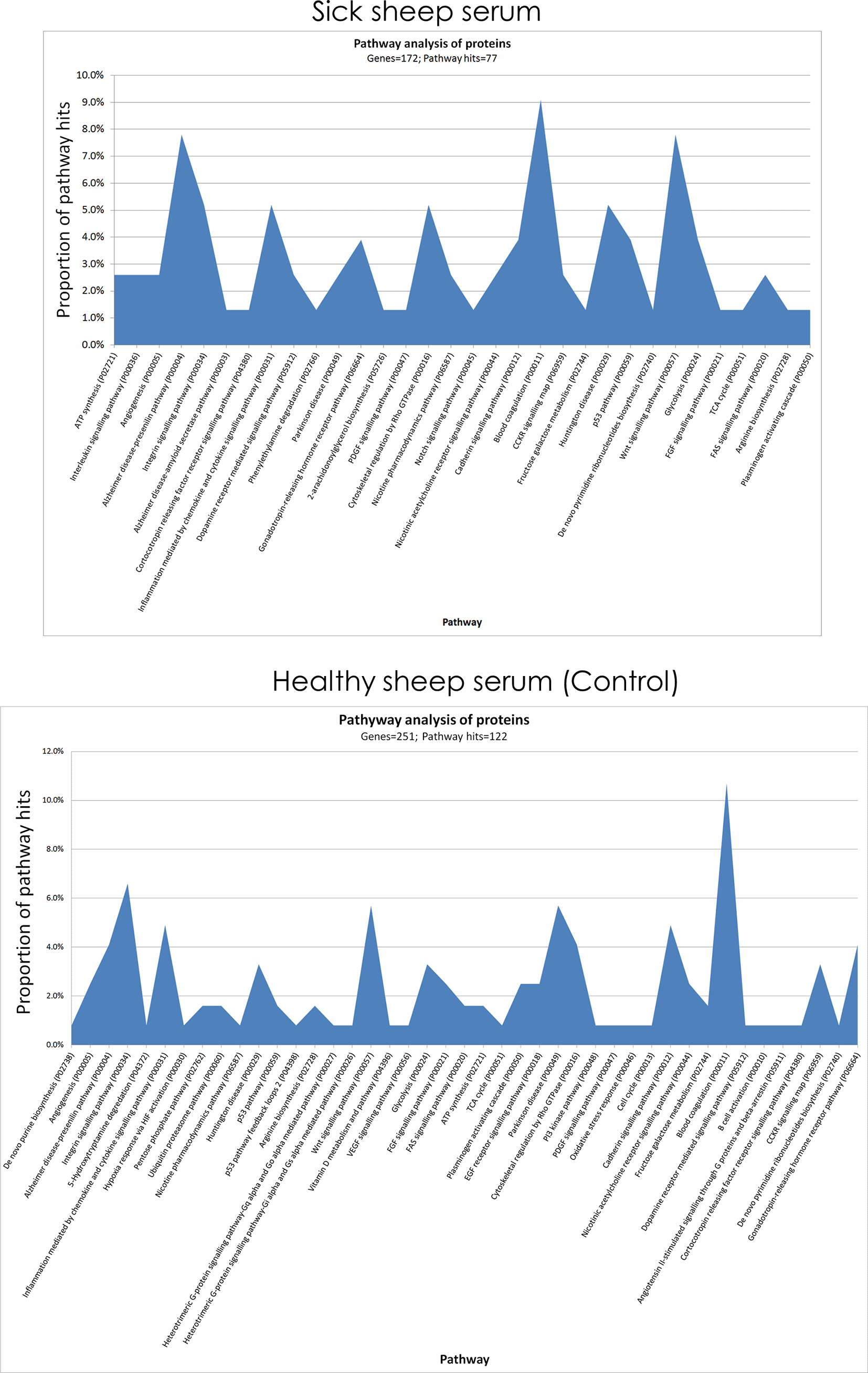
Note the differences in the enriched protein pathways between samples from sick and healthy sheep.
The sick sheep serum samples utilised for these experiments were submitted by referring veterinarians for analysis at UQ using automated methods for routine veterinary diagnostic work up. The samples were accompanied by brief presenting clinical case histories presented in Additional file 1.48 Follow-up information on these cases was not available in laboratory entries obtained for this report. With the clinical case history data at hand, the most important of them were signalment (except for two samples (SCi504 and SCi508)) whose ages were missing and the laboratory findings from the analysis of the samples.
The experiments for generating this report utilised SWATH-MS technology to primarily analyse proteins in ex-diagnostic serum samples of sheep suffering from a range of different ailments in comparison with serum from healthy sheep to detect any differences in their protein profiles. The strategy adopted involved testing and validating the potential application of the nascent peptide spectral library built from circulating acellular proteome of sheep.6 The overall intention here was to target protein analytes in significant laboratory findings of the submitted sheep serum samples based on the accompanying clinical data from UQ’s School of Veterinary Science laboratory. The relevant analytes for proteomics studies for this report are found in Additional file 1.48 Among the analytes of interest were gamma-glutamyl transferase (GGT), total protein (TP), serum albumin (Alb), aspartate aminotransferase (AST), alkaline phosphatase (ALP), creatine kinase (CK), fibrinogen and some other analytes that were listed in the reference ranges, for example globulin and serum amyloid. Another important analyte – at least in ruminants – that was not listed in the laboratory panel is haptoglobin.25 These analytes may have appeared as isoforms of their known protein moieties. In some cases however, the genes coding for these proteins were not well defined, possibly due to the consideration that the sheep genome still remains to be fully annotated.26,27 Nevertheless, it is feasible and practical to use draft genome sequences for the interpretation of MS/MS proteomics data without the need for tedious genome annotation.28 This does not negate the point that a well-annotated genome is necessary in order to name genes and proteins unambiguously.29–32
Differentially expressed serum proteins between healthy and sick sheep were either known or considered to be potential biomarker candidates for diagnosing and monitoring disease states, and this could also provide leads for further research endeavours. There are two important layers to determining the significance of the differences between sick and healthy sheep serum proteomes observed in this work: a) the degree of confidence in the observed difference from an analytical standpoint and, b) the biological plausibility that such differences truly correlate with a disease state. The discussion that follows looks at these preceding two aspects.
A problem of working with the present approach of identifying proteins is that when protein isoforms are not explicitly named and identified, it introduces ambiguity that makes it challenging to identify even well-known proteins as in the instances presented in this report. For example, in the set of samples utilised in this study, Hb was identified as A0A0F6YFJ0 (Beta-C globin). There were also two protein IDs representing GGT which comprised of W5QCX2 (Transglutaminase 1) and W5PB04 (Transglutaminase 3). Serum albumin was identified as W5PWE9 which is yet to be characterised for sheep in UniProtKB. It should be noted that one isoform of albumin, P14639 (Albumin), that was not identified in this set of experiments, has already been characterised in UniProtKB. The ID feature for Aspartate aminotransferase (AST or GOT) was W5PS88. There was no protein ID directly matching ALP in this dataset. However, there were three other phosphatases including W5P195 (Dual specificity phosphatase 14), W5P3B0 (Phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase) and W5QE45 (Serine/threonine-protein phosphatase) that were identified. Meanwhile, CK was represented by two IDs: W5PJ69 (Creatine kinase) and W5NQ67 (Creatine kinase). Fibrinogen was represented by four proteins including W5Q5H8 (Fibrinogen alpha chain), W5NQ46 (Fibrinogen beta chain), W5Q5A6 (Fibrinogen gamma chain) and W5PH03 (Fibrinogen like 1). Serum amyloid was represented by W5PJG0 (Serum amyloid A protein), P42819 (Serum amyloid A protein) and W5PJR0 (Serum amyloid A protein). Haptoglobin was identified as W5P0Q4 (Haptoglobin).
In veterinary clinical pathology, the determination of the quantity of protein in plasma or serum has always been traditionally based on the amount of albumin and globulin fractions,33,34 and yet there is more to the two broad protein groups when it comes to deep proteomic analysis. For example, in the present set of protein identifications, the globulin fraction was represented by at least 12 protein families or groups. The globulin protein fraction comprised of W5P812 (Protein AMBP), W5PSC1 (Ig-like domain-containing protein), W5Q0R1 (Sex hormone binding globulin), W5NQW4 (Alpha-1-macroglobulin-like), W5PGT9 (Ig mu chain C region), W5QI15 (Ig-like domain-containing protein), W5PSK4 (Ig-like domain-containing protein), A4ZVY9 (Beta-2-microglobulin) W5NSA6 (Alpha-2-macroglobulin), W5PPQ8 (Joining chain of multimeric IgA and IgM), P49920 (Corticosteroid-binding globulin) and the various VH region immunoglobulins such as A2P2G1 and A2P2I1. Being able to identify more proteins in the different fractions of serum proteins gives this approach an edge over traditional clinicopathological protein assays.
The experiments in this report utilised samples from single sheep case reports as proof-of-concept that SWATH-MS technology can be applied to identify vast numbers of proteins and their alterations in clinical samples of veterinary patients. The comparison of clinical cases (sick sheep serum), versus normalised serum from a large number of healthy sheep is a classical approach typical of biomarker discovery or detection studies.35 With this foundation in place, it is possible to establish a standard for routine proteomic analysis of serum samples submitted to laboratories in future, once a specific baseline serum proteome of sheep has been optimised using far much larger numbers of samples. This method also has the potential to be used for identifying different protein species that show differences between samples. The pooling of samples from healthy sheep was conducted on the premise that the pooled sample would provide a representative proteome of normal sheep serum. Similarly, the rationale of using a pooled sample from sick sheep followed the same premise that that this could provide a representative picture of all the proteins present in the sick sheep, particularly proteins that might only be abundant in specific disease states that could not be detectable in serum samples from healthy individuals. The inherent downside of pooling samples is that this strategy disregards the biological variation of the individuals the samples are drawn from,36 by capturing for example, the ‘average’ proteome profile across a population. And also in the present results, 26 protein IDs were not detected when samples from sick sheep were pooled, as opposed to them being analysed individually (Figure 2B). The reason behind this observation is not immediately clear, but batch effects during sample analysis could be advanced as a contributing factor,37 or possibly unknown effects of pooling samples for downstream analysis. It also follows that the use of pooled serum, at least from normal individuals to act as a control, is an accepted scientific practice for normalising samples that has also been widely used in some human studies.35,36
The evaluation of chromatographic features of peptides from samples – TICs in this case – was a practical, inexpensive and straightforward visual way of comparing ion intensities between small numbers of samples as previously reported elsewhere.38 Here, only two sample TICs were loaded per displayed panel on PeakView®: one from a sick sheep versus a normalised serum of normal sheep (Additional file 248). The TICs represented a measure of relative abundance of the peptides detected.39 It follows therefore that during interpretation of the TICs, it should be considered that they represented summed signals from the sample as well as background noise.40 The use of TICs in addition to SWATH-MS data extraction has a high depth of analysis whilst factoring in the wide dynamic range in analytes, is a useful strategy especially when considerable differences are expected between samples38 as in the present study. There were distinct differences between TIC profiles of sick sheep serum samples compared to serum profiles from normal sheep; this difference was more marked in the sample from the sheep with scabby mouth lesions (Additional file 2 48). In the majority of the analysed samples, the TIC intensities of sick sheep serum samples were generally higher than those of normal sheep serum, except for the ill-thrift and the ill lamb cases. A possible explanation for this observation could be malnutrition/starvation in the chronically ill sheep, since decreased food intake depletes protein generally and this was even more crucial in the neonatal sheep (ill lamb) that was likely still reliant on colostrum.41–43 Proteins in circulation are either derived from synthesis or from degradation, but considering that the small intestine is the most important organ for synthesis and absorption of proteins in ruminants,44 the detected proteins in starving sheep are therefore most likely to have been derived from tissue degradation. As a result, there was probably a comparatively very little protein reserve to elicit the higher protein intensities compared to the other relatively acute disease case samples. As for the analysis of the pooled sample of sick sheep (SCi511), the TIC profile was generally above that of pooled normal serum up to approximately 38 min when it switched below the pooled normal serum TIC, before peaking again at the hydrophobic end of the chromatogram. An explanation for this observation remains to be established and is open to further interpretation.
The differences in protein species between individual serum samples from sick sheep could have been due to the different aetiological and pathological factors of the presenting condition in the different sheep that could have stimulated the production of different proteins or their alterations. Without follow-up clinical data on the cases or a definitive diagnosis, it is not possible to fully determine and to explain these differences. Also, clinical cases were from diverse populations which may have contributed to different serum proteomic profiles as proteomes are known to be dynamic.45
Different numbers of proteins were identified from the different samples and to some extent, different protein species. The sample from the sheep with scabby foot and mouth lesions had the highest number of protein IDs (Figure 2A). A substantial number of protein IDs were common between sick and the normalised sample from healthy sheep as represented by the ‘pooled healthy control and sick overlap’ legend item in Figure 2A. The protein yield of 210 IDs from the pooled sick sheep sample (SCi511) was much lower than expected as compared to the IDs from all other individual samples considered together. It is not immediately evident as to why this was the case. This relatively low numbers of protein IDs could potentially have been due to sub-optimal tryptic digestion of this particular pooled sample, loss of peptides during sample preparation or probably due to unknown factors that influence the pooling of samples.
There were differences in protein intensities between the samples as evident in the ten most abundant proteins in each analytical case (Figure 3). The comparison of protein intensities of the pooled serum from healthy sheep (SCi512) versus pooled serum from sick sheep (SCi511) revealed considerable differences for some proteins, with each of the points on the graph representing a protein (Figure 4A). Fewer proteins (210 IDs) were identified in sick sheep sample pool, as compared to 335 IDs in healthy sheep serum pool. It should have also been possible to have a relative quantitation of the proteins centred on their intensities benchmarked on the calibration of the intensities from healthy sheep serum based on the principle of area under curve of the TICs46,47 (Additional file 248). Using this approach, could have demonstrated the feasibility of determining what proteins were either upregulated or downregulated in the clinical serum samples from sick sheep.
The GO term classification provided an overall picture of the identified proteins in both sick and healthy sheep serum (Figure 5). An important observation from the protein pathway analysis is that it provided 11 clearer protein pathways that peaked in sick sheep serum (Figure 6). The roles of some of these protein pathways in sheep remain to be determined, but they have been studied considerably in humans and other model species such as mice. It is not unreasonable therefore to suggest that with homology, there is a translational potential in the observations made in this study from sick sheep that can be learnt from.
There were limitations in this experimental study. The first one was that there was only one biological sample for each clinical case study — which is not unusual for case studies. Since each clinical case was unique, to be able to adequately test current observations, three or more technical replicates (or MS injections for that matter) out of each biological sample would have improved the meaningfulness of the data score plot. The second limitation was that it was not possible to re-analyse samples drawn from the same sheep after they recovered (i.e. to act as their own controls), and/or at different stages on the disease to determine if chronicity had an effect on protein species or their alteration – considering that sick sheep samples were opportunistically utilised from laboratory archives at UQ.
The use of SWATH-MS analysis successfully identified proteins and enabled protein profiles from serum samples of sick and healthy sheep to be distinguished. It was possible to detect some established clinical biochemical analytes, for example aspartate aminotransferase, creatinine kinase, haemoglobin, serum amyloid A, fibrinogen, haptoglogin, members of gamma-glutamyl transferase group, serum albumin and various proteins of the globulin fraction. Protein pathway analysis provided useful information on the expression profiles of protein groups between sick and healthy sheep by giving them a biological meaning. There was a downside to this approach, for example the detection of alkaline phosphatase – an important analyte that was not obviously identifiable in this dataset. Some detected proteins of sheep such haemoglobin, haptoglobin and some isoforms of serum amyloid did not have distinct gene names as compared to homologous counterparts in related species. Nevertheless, this proof-of-concept study has demonstrated that with a known benchmark or standard samples from healthy individuals, it is feasible to determine relative protein alterations in clinical samples. This approach could have a place in veterinary clinical pathology in future.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)