Keywords
enterococcus, transposons, plasmids, antimicrobial resistance, blast, data indexing, bioinformatics, data mining, metadata, metagenomics, algorithmic information
This article is included in the Bioinformatics gateway.
This article is included in the Pathogens gateway.
This article is included in the Hackathons collection.
This article is included in the Antimicrobial Resistance collection.
enterococcus, transposons, plasmids, antimicrobial resistance, blast, data indexing, bioinformatics, data mining, metadata, metagenomics, algorithmic information
A variety of publications have reported the diverse mechanisms of microbial virulence (Leshem et al. 2020; Tarsillo and Priefer 2020; Nogueira et al. 2019; Saeki et al. 2020; Lange et al. 2019; Saiardi et al. 2018; Russo and Marr 2019; Feßler et al. 2018; Geisinger and Isberg 2017; Schroeder et al. 2017; Diard and Hardt 2017). In the vast majority of cases, one or more features/biomarkers can be identified and compared against databases of known virulence factors. Examples of such features can include genetic (e.g. pathogenicity islands, antimicrobial resistance (AMR) genes, mobile elements) (Kaushik et al. 2018; Vestergaard et al. 2019; Madec et al. 2017) and metabolomic (e.g. carbohydrates, lipids, small molecules) (Jia et al. 2019; Lloyd-Price et al. 2019) features.
The National Center for Biotechnology and Information (NCBI) at the National Institutes of Health hosts in-person hackathons to crowd-source the rapid development and prototyping of various methods (Connor et al. 2019). Recently (July 2019), a hackathon was convened to develop methods to detect various aspects of microbial virulence. The pipelines and methods that were generated during this hackathon were made publicly available to rapidly disseminate these tools and to enable the rapid identification of various microbial virulence factors.
The aim of this study was to develop computational pipelines that enabled cloud-based analyses to identify markers of microbial virulence in a modular and publicly available way. Attendees focused on implementing proof-of-concept strategies and methods to address “high-priority” needs. These tasks included data indexing, metabolic functions, virulence factors, AMR, mobile element detection in enterococci, and metatranscriptomics. Participants were grouped into teams, with each team developing at least one automated pipeline for their assigned task. These pipelines and workflows can be adapted by the research community to enable improved detection of various microbial virulence factors in a variety of data types.
All data and metadata from NCBI Sequence Read Archive (SRA) were assembled into contigs using SKESA version 2.3.0 (Souvorov et al. 2018). Virulence factors, AMR genes, and mobile elements were then identified and annotated in these contigs with AMRfinder (Feldgarden et al. 2019). These data and metadata were then uploaded to storage buckets and Big Query (respectively) in the Google Cloud Platform. A table containing pointers to the above data and metadata was then generated.
A script was then used to: 1) concatenate the metadata for each SRR in a subset of SRA experiments, 2) concatenate all assembled contigs for each SRR in the same subset of SRA experiments, and 3) apply a previously reported quantitative clustering by compression method (Cilibrasi and Vitanyi 2005), to identify any relationship between the complexity in contig sequence data and the associated metadata.
BioProjects within the NCBI-hosted Sequence Read Archive were selected for analysis based upon the presence of shotgun sequencing metagenomic samples from the gut microbiome and paired labels identifying the sample with a health status. Disease statuses encompass individuals diagnosed with colorectal cancer, diabetes, obesity, Parkinson’s disease, and ulcerative colitis. Metagenomic gut samples from healthy individuals were also selected. A subset of 100 representative SRA accessions (Table 1) were selected for an initial characterization to prototype the metabolic functions workflow. Sequence read files for the selected SRA accessions were downloaded using SRA Toolkit version 2.9.6, followed by a read quality filter using PRINSEQ++ version 1.2 (Schmieder and Edwards 2011). All filtered reads contained paired ends, which were concatenated into a single read file using a custom script before being passed as input into HUMAnN2 version 2.8.1 using the UniRef90 full database to optimize the identification of uncharacterized proteins (Franzosa et al. 2018). Following the HUMAnN2 workflow, output files were normalized based on relative abundance followed by joining all sample tables to create a single table for gene families, path coverage, and path abundance. All three tables were preprocessed using a custom R script before being passed through a random forest classifier. A total of 100 trees were generated for the binary classification using the random forest algorithm implemented in the Python scikit-learn library. Owing to the low sample size with 18 healthy and 41 diseased samples (belonging to Parkinson's disease, obesity etc.), leave-one out cross validation was used.
Experimentally validated virulence factor (VF) genes from the Virulence Factor Database (VFDB) were used to represent virulence-associated genes (Chen et al. 2016). Example metagenomes used for testing were drawn from public datasets listed on NCBI SRA and included healthy and disease-state human skin metagenomic samples (Table 1). Specifically, diseased metagenomes were drawn from the Diabetic Foot Ulcer metagenome study (BioProject: PRJNA506988) and healthy foot skin metagenomes were drawn from BioProject: PRJEB30094. Metagenomes were assembled using MetaSPADES version 3.13.1 (Nurk et al. 2017).
A Hidden Markov Model (HMM) was applied to the VFDB genes to create virulence profiles. Genes were selected for which at least five different bacterial species were available. Multiple sequence alignments were generated using MUSCLE (Edgar 2004) and HMMs using HMMER3 (Eddy 2011). Genomes and/or corresponding protein coding sequences were screened with HMMSEARCH using pre-computed significance scores. Scores were calculated as 80% of the envelope alignment score of a representative sequence corresponding to its HMM. Alignments were filtered by custom scripts to extract putative virulence factors’ loci. VF sequences were concatenated, aligned and used as input for phylogenetic analyses. Phylogenetic trees were constructed using RAXML-ng (Kozlov et al. 2019) and analyzed using the R package Ape (Paradis and Schliep 2019) and Newick Utilities (Junier and Zdobnov 2010). Virulence tags were assigned based on the number of virulence loci found and phylogenetic classification. All analyses are described within a Snakemake workflow (Köster and Rahmann 2018).
A support vector machine (SVM) model was also developed to classify virulent and non-virulent gene segments by training on a reference set of labelled pathogen and commensal genomes. The pathogen genomes were acquired from an NCBI Assembly search and included the species identified in the VFDB dataset. Commensal genomes were also acquired from an NCBI Assembly search, and included species selected from the NHSN Common Commensals List and from prior work (Busby et al. 2013). To characterize the novel virulence factors, the known virulence factors were masked from pathogenic genomes and non-pathogenic genomes.
Utilizing FASTQ files, either by providing them directly or by a list of SRA run accession numbers, this Nextflow pipeline creates a curated list of AMR genes present in a metagenomic sample and includes their context by performing a guided assembly with the detected genes. In this case, the sample was from a preterm infant (ERR1600439) with information of the context of the detected gene. After quality control, three modes to detect AMR genes are available: “magicblast, “hmm” and “mash”, all utilizing the AMRFinder AMR database (Feldgarden et al. 2019). The first mode utilizes magicBLAST to align the sequencing data to the reference database and, after filtering by coverage breadth and depth, outputs the selected AMR genes found in the sample. The “hmm” mode takes the sequencing data and translates it into the six possible reading frames, and breaks each into open reading frames (ORFs). These ORFs are used as input to the HMMER algorithm to search against the profile HMMs from AMRFinder and selects AMR sequences based on a threshold bit score. The final mode, “mash”, builds a MASH sketch with the same AMR database and screens the reads against the sketch. This produces k-mer distances between the read set and each AMR, which then is used to extract only the AMRs that are close to the readset. The resulting AMRs, from any mode of the pipeline, are used as bait to perform a guided assembly with SKESA version 2.3.0.
All 15,000 available E. faecalis and E. faecium sequence runs were downloaded from the SRA database. Short reads were assembled using SKESA version 2.3.0 (Souvorov et al. 2018), with AMRFinder used to identify resistance genes (Feldgarden et al. 2019). Genomes were annotated using the Prokaryotic Genome Annotation Pipeline version 4.8 (Tatusova et al. 2016). BLAST (Altschul et al. 1990) was used to compare the assembled sequences against a database of transposon and plasmid sequences. Annotations were also searched for terms associated with mobile elements, including transposase, recombinase, integrase. After performing the described BLAST analyses, a report that combined and summarized these results was constructed.
Processed data was downloaded from the Inflammatory Bowel Disease Multi'omics Database (IBDMDB; PRJNA398089). Patient metadata, including inflammatory bowel disease information, was also obtained from the same source. Metagenomics and metatranscriptomics data were obtained as taxon and pathway abundance tables, respectively. Comparison of metagenomic and metatranscriptomic abundances were done using custom code.
To see whether metatranscriptomic data is predictive of disease state of an individual, seven machine learning classification models were trained and subsequently combined in a consensus model that classifies disease outcome. A random selection of a third of the individuals were placed in a test set to evaluate the performance of the consensus model and these individuals were not used in any training step. The input features consisted of the relative abundance of each metabolic pathway, which were normalized to have 0 mean and equal variance. The models trained include Decision Tree, K-Nearest Neighbor, Support Vector Machine, Logistic Regression, Random Forest, and Multi-Layered Perceptron. Each model was trained on the training data, using hyper-parameter tuning with five-fold cross-validation. The seven models were then combined using a majority vote scheme into a consensus model. The performance of the consensus was evaluated on the training and previously unseen test datasets. All machine learning analysis was done using the Python library scikit-learn.
Before beginning the hackathon, we assembled the relevant sequence data from SRA into contigs prior to annotating them with known virulence factors, AMR genes, and mobile elements (for Enterococcus-related samples). We then added these processed data, together with the original SRA metadata and the pre-computed virulence metadata into the Google Cloud Platform. These data and metadata formed the basis for the downstream analyses described below.
The data indexing group focused on the underlying requirement by funding agencies to submit the experimental data, together with the associated metadata, to public data repositories. These large repositories include an extremely large corpus from which to perform data mining activities on existing data. Such activities are enhanced when the submissions conform to the findable, accessible, interoperable, and reusable (FAIR) data principles (Reiser et al. 2018). While the repositories provide a search interface that enables filtering by user-specified criteria, searching on relationships between datasets and supporting downstream data mining activities are often not supported. As such, the data included in this hackathon were all hosted in the cloud, using Big Query within the Google Cloud Platform. This approach facilitates the rapid identification of relevant datasets based on not only SRA metadata, but also using preprocessed data and metadata including assembled contigs, presence of AMR genes, and other elements in the contigs. Providing community access to this, and other relevant information in the cloud will improve the pace of developing solutions for human health.
The aim of this effort was to measure the feasibility of storing the assembled contigs, original SRA metadata, and additional metadata in a cloud-based platform to facilitate downstream analytical workflows (Figure 1). The benefit of this design is that both the tools and the data are co-hosted in the same cloud environment. This approach minimizes time required for file transfer and ensures that workflows are consistently accessible to all users.
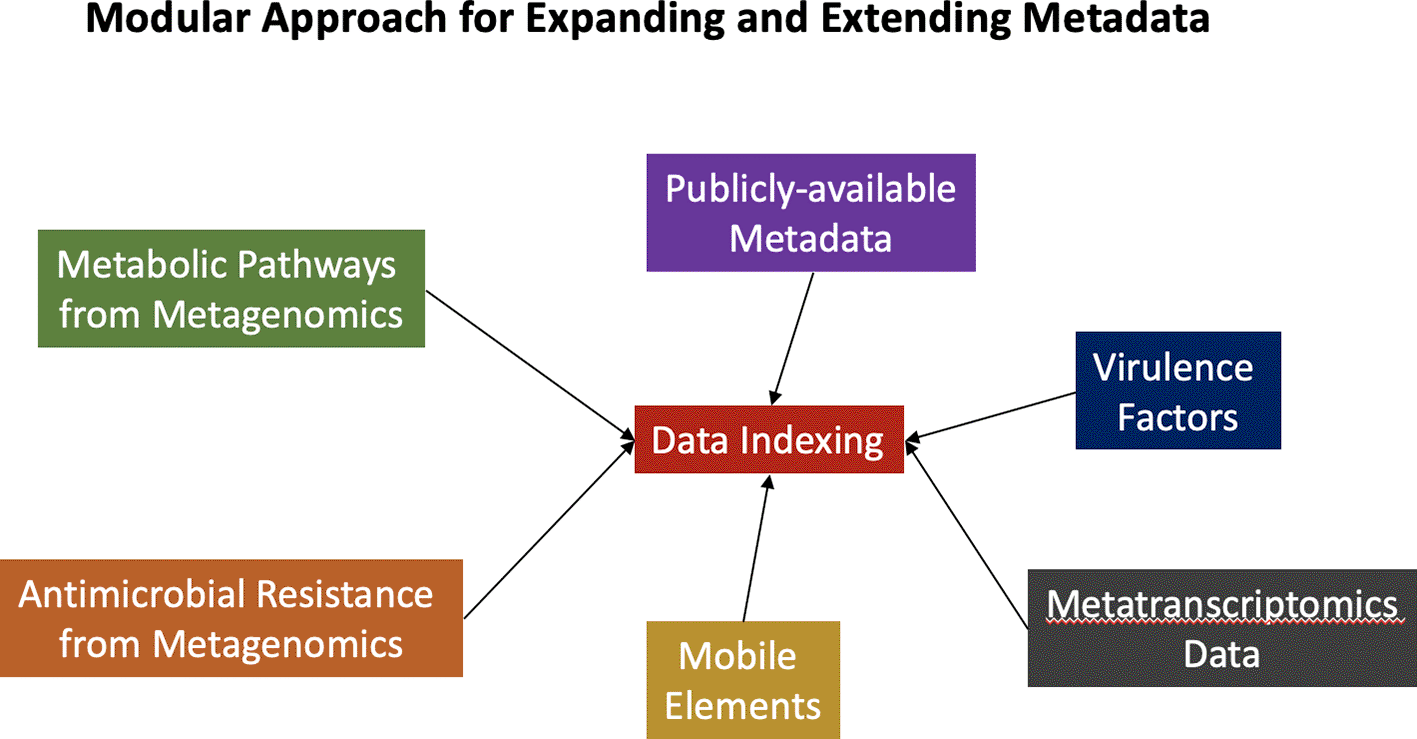
A graphical representation showing the important relationship between data generation, data analysis, and data indexing.
In addition to the storage of data, we hypothesized that the complexity, or sequence diversity, of contigs could correlate with the complexity of the associated metadata (e.g. disease state, year, host species, AMR gene presence). To quantitatively compare metadata and sequence data we used a technique based on algorithmic information theory. Briefly, this approach takes advantage of the fact that two related sequences show a high degree of sequence similarity (i.e. low genetic distance), and the files containing this information could therefore be compressed more efficiently. Similarly, files containing metadata that is highly repetitive would also compress much more efficiently. As such, we applied a previously published metric of mutual information that compares how well sequences and metadata compress individually and how well they compress when concatenated together (Cilibrasi and Vitanyi 2005) (Figure 2). This algorithm was shown to be agnostic to the input data and consequently robust in a variety of applications including DNA sequences. Their previous study showed that known biological taxonomies could be replicated without having to adjust the metric. A related paper demonstrated that comparing the dictionary sequences compared to the logarithmic size of the dictionary, it is possible to identify statistically significant sequences by length above a threshold (Milosavljević and Jurka 1993). Applying this method to pairs of SRR identifiers from SRA and their associated metadata revealed better compression of the metadata overall than the sequence data. We expect that the combination of more detailed and descriptive metadata for future SRA submissions, together with generating additional metadata from the relevant sequence data will improve the performance of this analytical method.
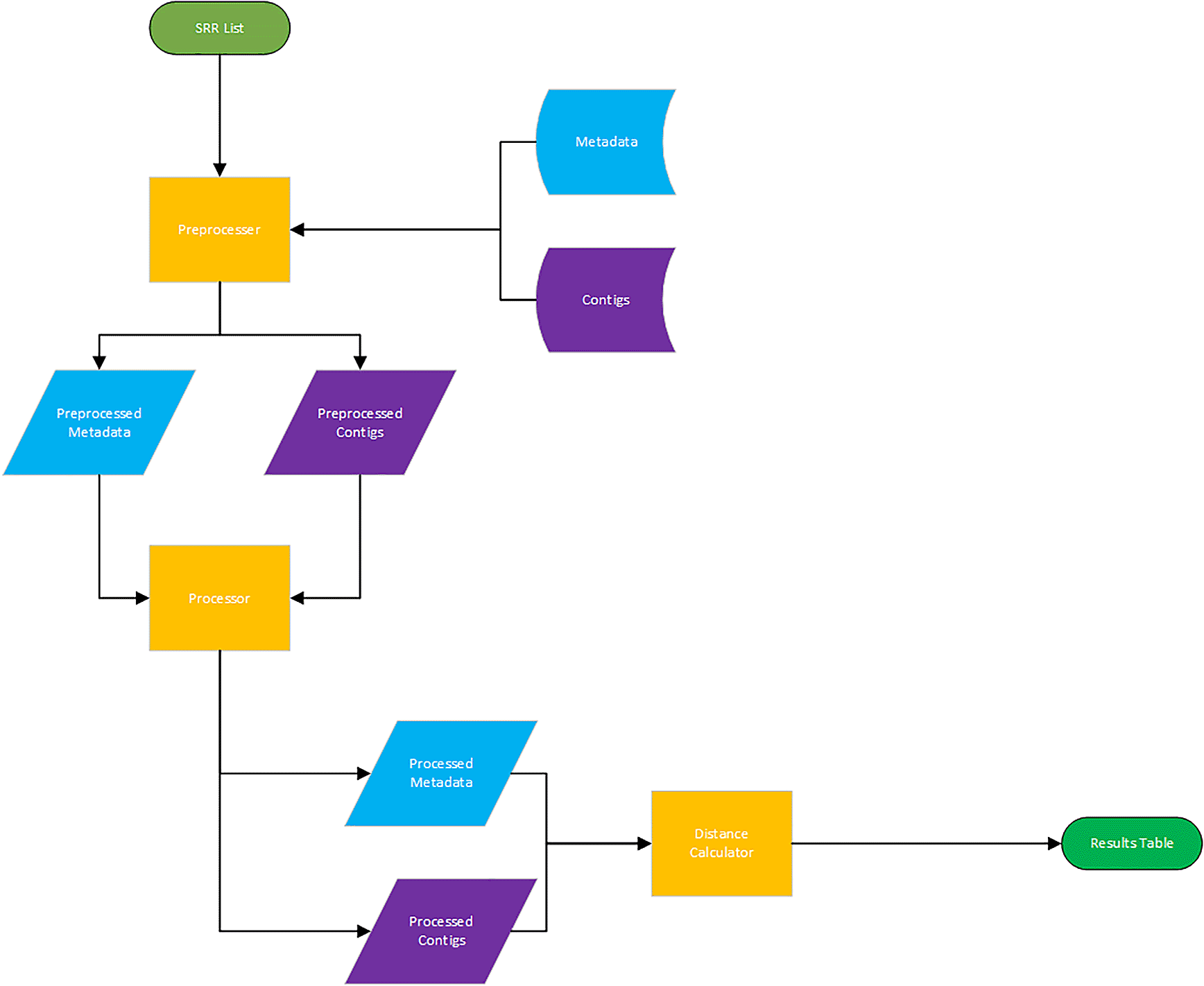
The computational workflow that was used to test whether sequence complexity in contigs relates to metadata complexity.
The metabolic functions group focused on identifying metabolic signatures of relevant virulence factors from a subset of the approximately one million metagenomic datasets that have been deposited at NCBI in the past two decades. Tens of thousands of these sequences were human-related. Due to the diversity in the metabolic capacity of the human microbiome, the role of the gut microbiome has been extensively studied in metabolic disorders and chronic diseases. At the same time, the value of machine learning has been recognized and applied to study complex datasets such as next-generation sequencing datasets. The goal of this effort was to uncover the missing metabolic functions that could exacerbate disease or disorder conditions. Finally, the established model would help predict the gut microbiome metabolic profile that may be associated with dysbiosis in the human body.
The goal of this project was to identify unique metabolic functions in gut metagenomes for disease states such as colorectal cancer, diabetic, obese, Parkinson's disease, and ulcerative colitis populations. We began by retrieving a variety of publicly available human samples from the SRA. These samples were derived from patients with a variety of disease states and were subjected to a novel computational workflow that transforms metagenomics sequencing reads into metabolic profiles (Figure 3). Specifically, we generated 100 trees in the random forest to select the features with the depth of 2 for pruning and classification. We then calculated the useful performance metrics for this approach including precision (1.00, 0.72), recall (0.11, 1.00), and F1-score (0.20, 0.84) for non-disease vs disease samples respectively, along with the area under the curve value of 0.86 (Figure 4). Overall, we observe that this cloud-based workflow shows a high amount of promise for differentiating between disease and non-disease samples using only metagenomic data.
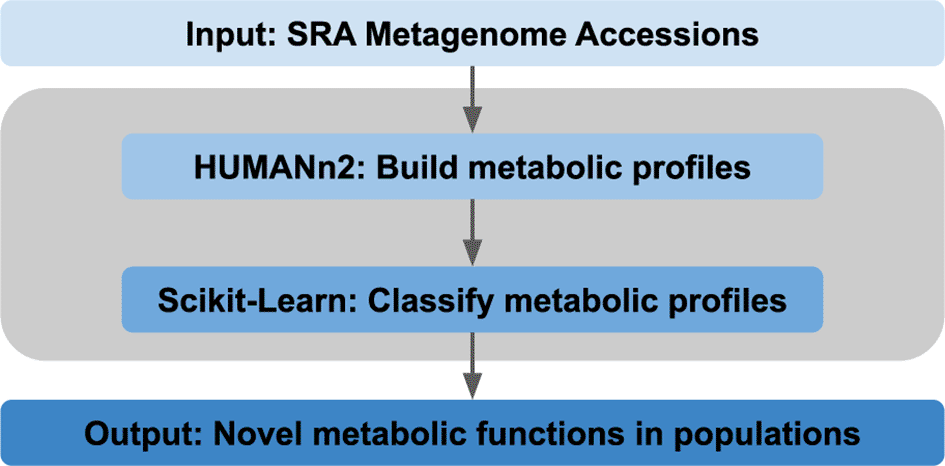
A graphical description of the computational workflow used to identify metabolic functions from metagenomics data.
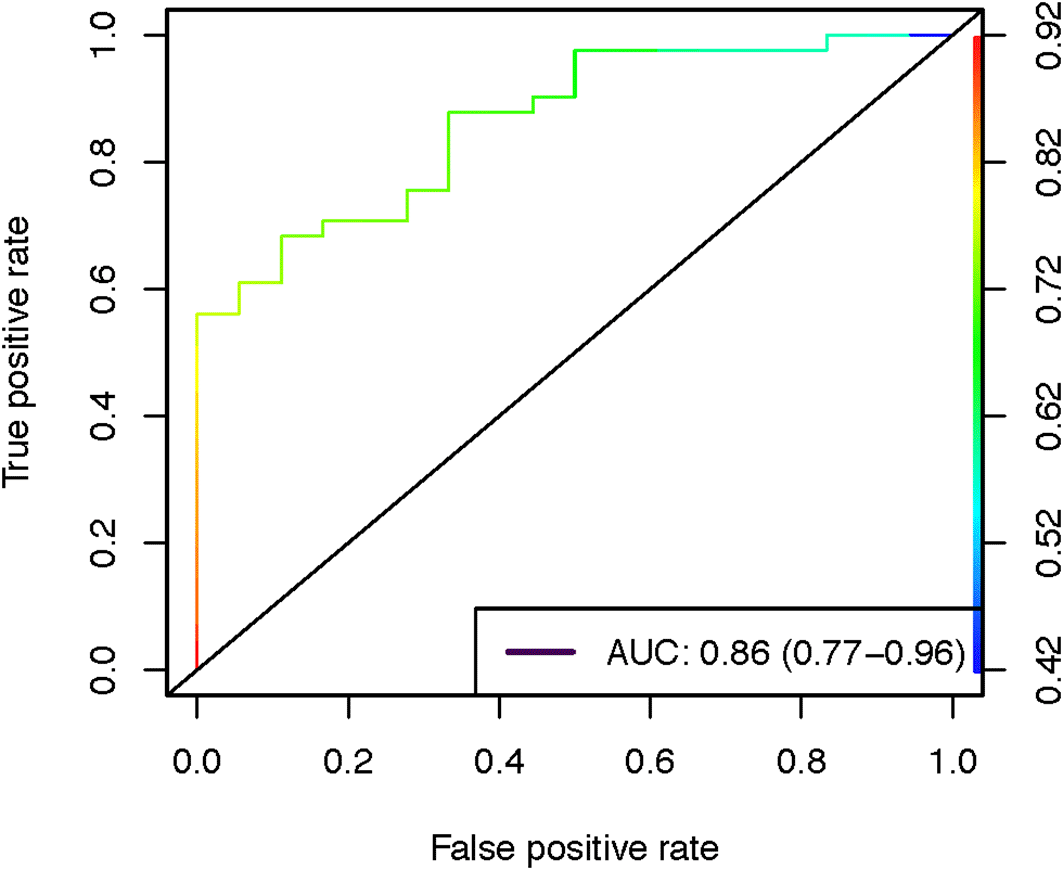
AUC quantifies the ability of the machine-learning based process to correctly assign healthy and unhealthy patients based on metagenomics data.
The virulence factors group developed methods to better understand how disease progression could be impacted by: metagenomic identification of microbial virulence, community relationships, and environmental context. Maintaining an appropriate scope for the identification of new virulence factors can often be challenging. Especially since certain contexts, such as the introduction of at least one virulence factor in a population can significantly alter the landscape of the surrounding microbiome and the pathological potential of individual microbes, including those normally considered commensal. Crucially, few tools exist for probing microbial virulence in metagenomes, which constrains the development of culture-free methods in public health and epidemiology.
We found it difficult to manually identify and acquire datasets with sufficiently detailed metadata for this task. After identifying an appropriate metagenomic study, the data from both healthy and diseased human skin microbiomes was subjected to the HMM portion of the workflow (Figure 5). We found that assembling the sequencing reads was relatively fast but was still the limiting factor in the overall speed of the analysis. Although the workflow that we implemented seems logical, additional software improvements are necessary to improve tractability and obtain the desired results in a reasonable amount of time.
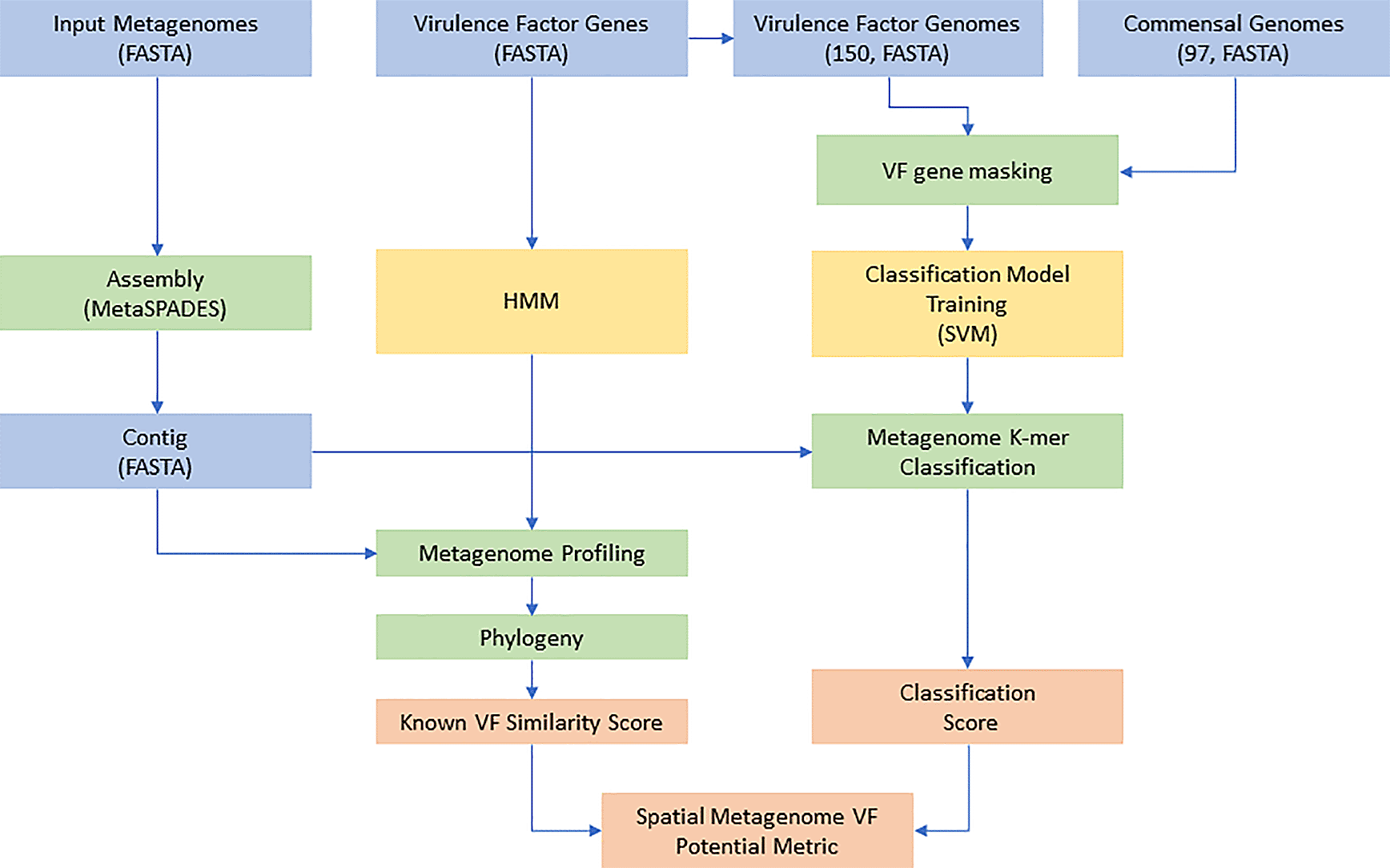
A graphical representation of the workflow to predict the presence of virulence factors from metagenomic datasets.
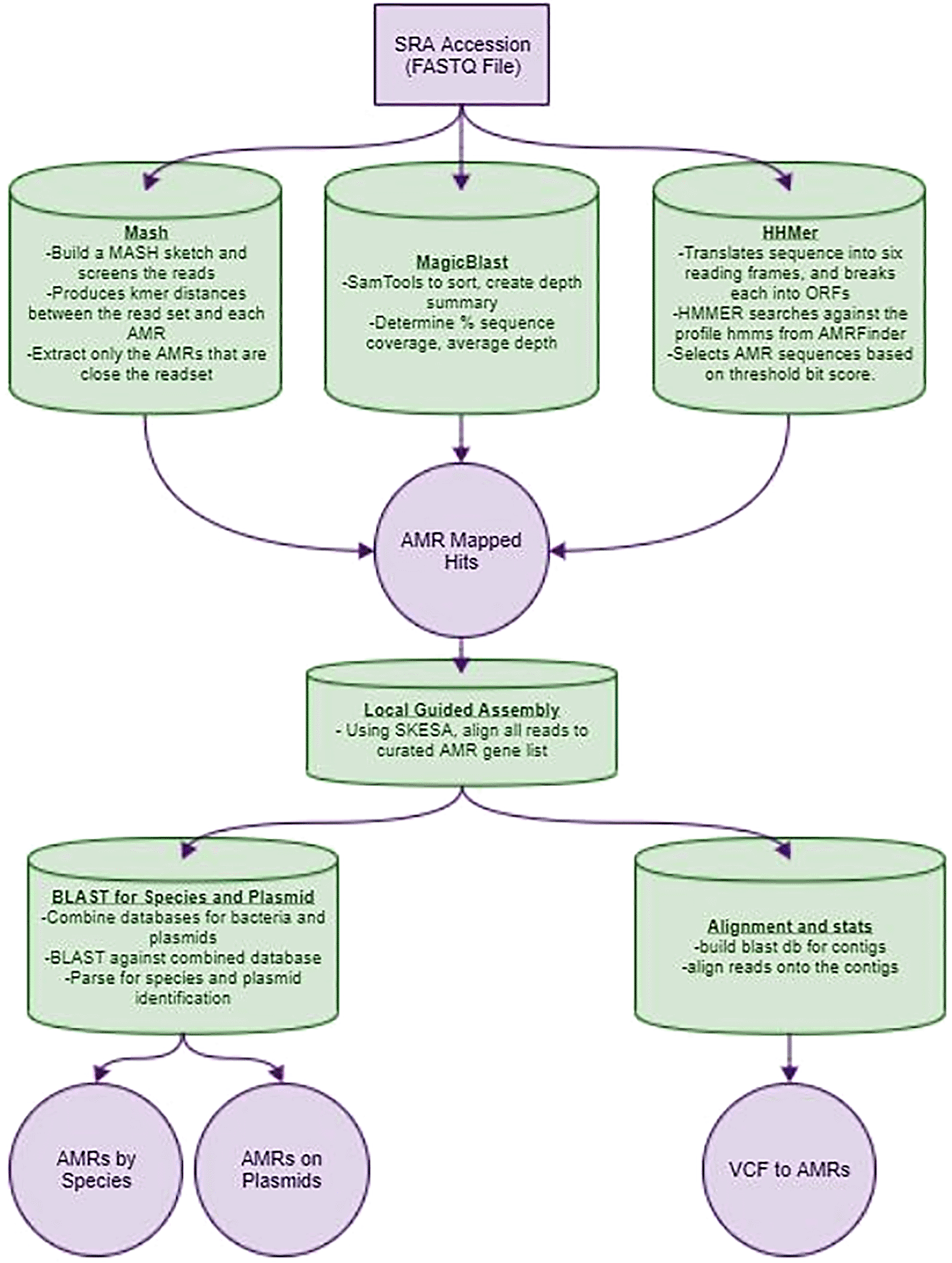
A depiction of the computational workflow used to identify antimicrobial resistance markers in metagenomic datasets.
AMR has been identified as a global concern, as microorganisms worldwide gain the ability to resist drugs that have been used to treat common infections. Identifying AMR genes has been the focus of several organizations, leading to the curation of large bacteria-focused databases such as the Bacterial Antimicrobial Resistance Reference Gene Database, and a call for the characterization of these genes (Buckner et al. 2018). Plasmids, which are self-replicating elements, can contain AMR genes and as they are often transmissible through horizontal gene transfer, have become a concern for the global spread of antibiotic resistance (San Millan 2018). Given that a human microbiome sample includes thousands of species, surveillance and the detection of AMR genes is critical. Developing tools that can not only identify which AMR genes are present but that can also classify which bacterial species the AMR genes are likely from, and whether these AMR genes were likely derived from a plasmid, is extremely informative. The intersection of this information is of utmost importance as plasmids have been identified in the spread of AMR in specific bacterial families, such as Enterobacteriaceae and Enterococcaceae.
We designed each of the pipelines such that they generated a list of AMR gene “hits” from the sequencing reads used as input. In this case we used the publicly available human fecal gut SRA sample ERR1600439, available from NCBI. We observed that the number of hits varied depending on the algorithm that was used. Specifically, we found that magicBLAST identified 17 AMR candidates and MASH identified 45 AMR candidates. After guided assembly, each pipeline created a varied number of contigs, with magicblast creating seven, which was the most generated by any of the three algorithms.
Infections caused by Enterococcus spp. present a major threat to public health as a leading cause of nosocomial infections. One major contributor to the severity of infections caused by enterococci is its frequent resistance to antimicrobial therapy. The association of resistance with mobile elements is of particular concern, as this can result in the rapid spread of AMR to previously susceptible isolates. Thus, the development of tools to identify mobile elements and their association with resistance genes will fill an important gap in current knowledge and provide a useful resource for public health and research applications.
Our mobile element (“mobilome”) analysis was implemented as a computational workflow capable of identifying antimicrobial resistance genes that are flanked by active mobile elements (Figure 7). The tabular report that was generated by the workflow consisted of rows that contained information about each isolate-feature combination. The location, feature type, analysis method, name, and relevant BLAST statistics for each isolate-feature combination are reported in a tab-delimited text format. BLAST was used to compare each individual genome against each reference plasmid to determine if a particular reference plasmid is present in the sequence of a given isolate. The global coverage of each plasmid by each isolate is reported in a tab-delimited text format.
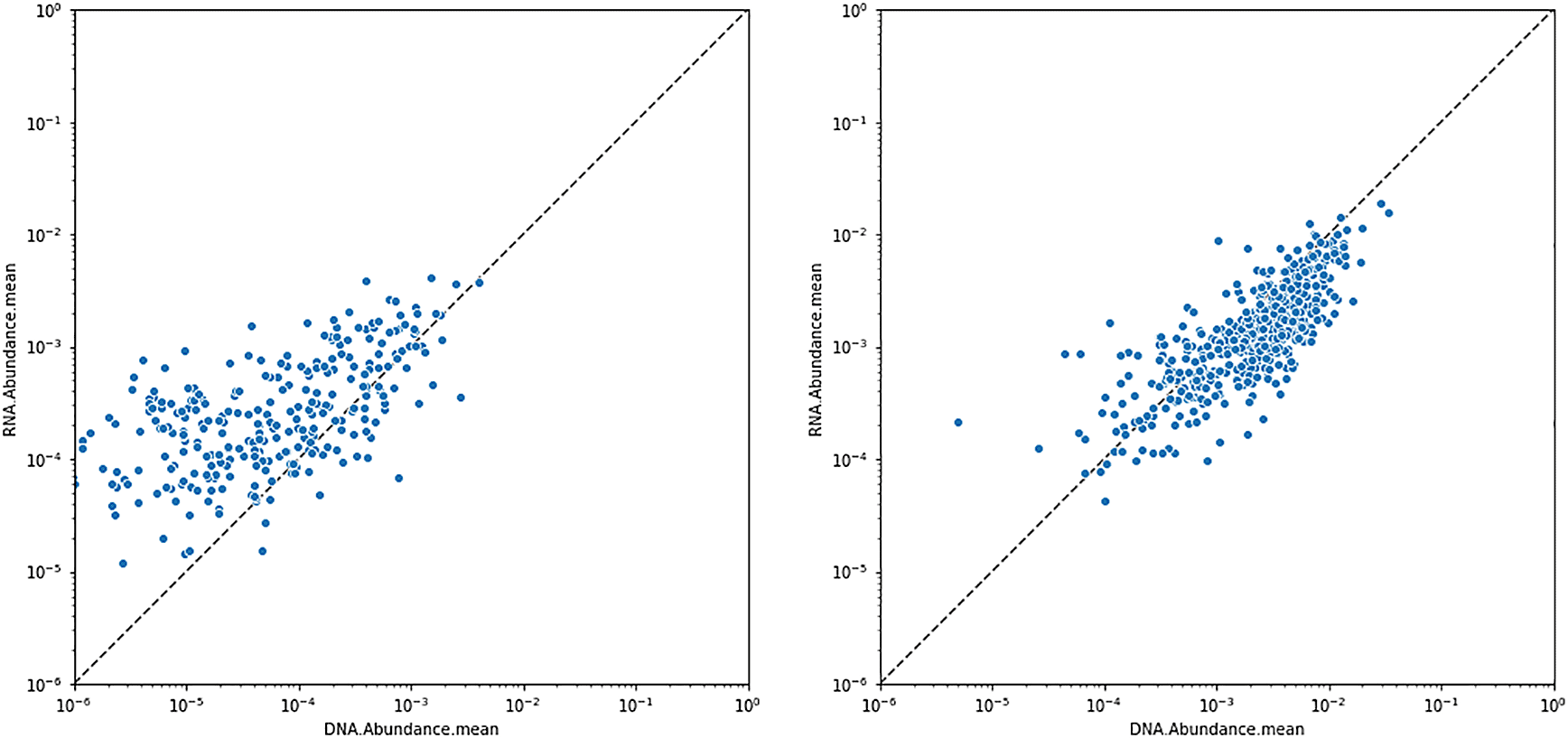
Mean RNA and DNA abundance values for Eschericia coli (left) and Bacteroides vulgatus (right). Each point represents a patient sample. Most patients have lower (relative) amounts of RNA from B. vulgatus compared to DNA; and the converse is true for E. coli.
Use cases
We wanted to construct a pilot workflow within this cloud-based platform to rapidly and effectively preprocess and analyze metatranscriptomic data. This workflow could be used to support future analyses to distinguish between active and inactive/dormant bacterial taxa in a given sample. The aim for this use case was to determine whether we could detect such differences in bacterial populations using metatranscriptomics, and whether any biological insight could be derived from the results. While many interesting questions can be pursued by analyzing metatranscriptomic data, we decided to quantify differences between levels of DNA and RNA for a given gene--to distinguish between persistence and active metabolism.
Persisters are often defined as metabolically inactive bacteria, characterized by arrested growth, low ATP levels, and low mRNA expression. Bacteria can enter a persistent state when exposed to supra-lethal concentrations of antimicrobial compounds, and can resume growth after the compound is removed from the environment (Fisher et al. 2017). Thus, persistence is a form of transient antibiotic tolerance, and can result in treatment failure and relapse of infection in the clinic (even in the absence of antibiotic resistance). It is therefore crucial to be able to detect persisters in clinical samples. One possible method of detection of persisters is genome-wide absence of RNA transcripts, but a presence of genomic DNA. This can be accomplished by considering the mean or median RNA vs DNA abundances across all pathways of each individual species (Figure 7).
Similar to how a human cell with one genome can differentiate into vastly diverse cell types, similar metagenomic compositions in microbial communities can have vastly different transcriptional/functional profiles (Franzosa et al. 2014). Many genes/pathways demonstrate a strong correlation between RNA and DNA abundance. However, there are also examples of pathways where DNA abundance does not vary much across samples, but RNA abundance does (Figure 8).
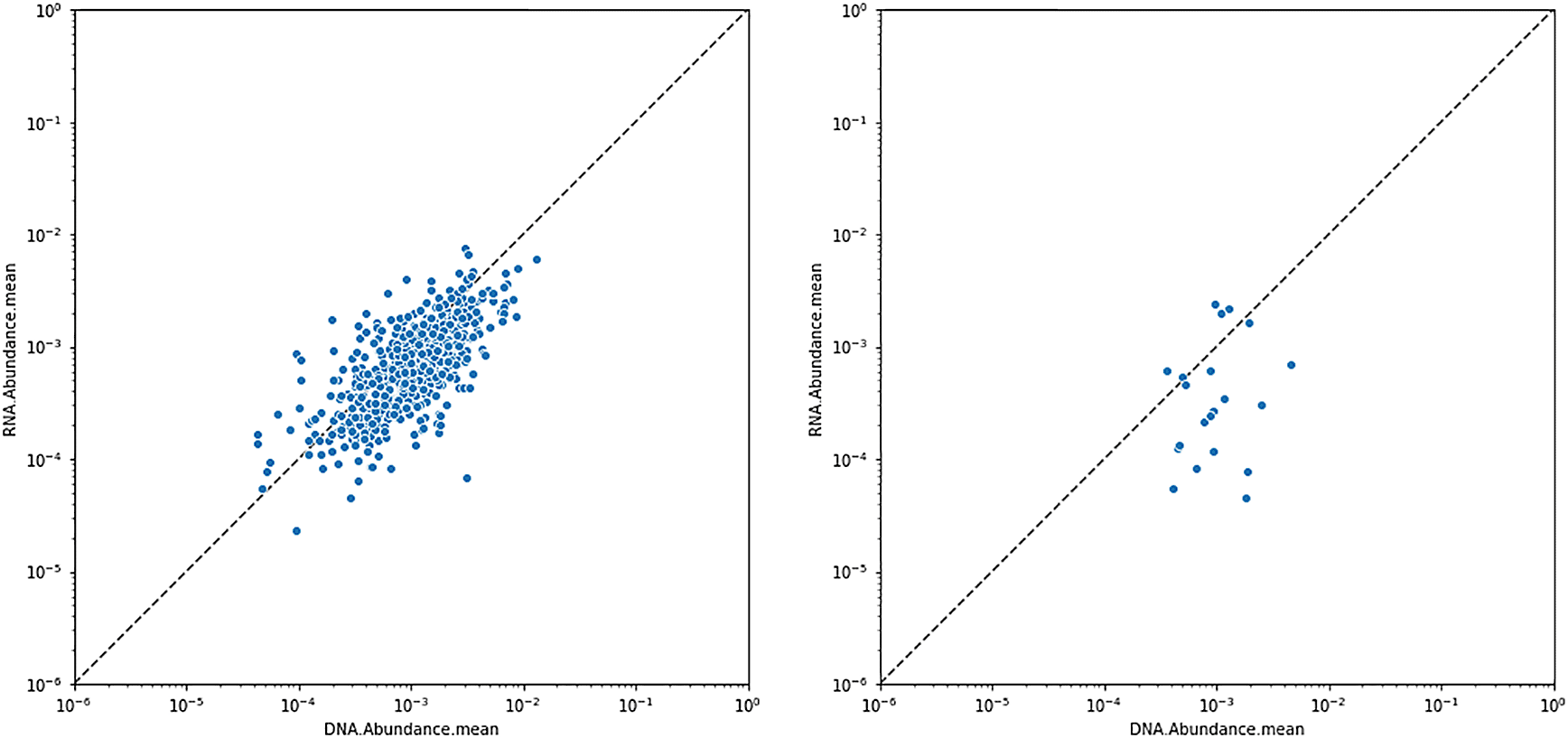
Arginine biosynthesis (left) and Fucose-rhamalose catabolism (right) pathways. Each point is a patient sample. For the arginine synthesis pathway, RNA and DNA abundance correlate well. However, for the fucose-rhamalose catabolism pathway is an example of low variability in terms of DNA abundance, but high variability in RNA abundance.
Recent studies classified healthy vs diseased patients based on NGS data (Bang et al. 2019) (Pasolli et al. 2016). Early analysis of metatranscriptomic data has identified differentially-expressed pathways in periodontal disease when compared to healthy oral microbial communities (Jorth et al. 2014). Metatranscriptomic profiling data potentially has richer and more informative features than metagenomic or 16S data; since it provides information on which genes and pathways are being actively expressed, which may contribute to a host phenotype. We therefore selected the IBDMDB dataset for training ML models since it has metatranscriptomics data, and disease metadata.
As a pilot experiment, we trained seven machine learning models (decision tree, logistic regression, SVM, Naive Bayes, K-nearest neighbor, random forest and multi-layer perceptron) using the pathway abundance data extracted from the metatranscriptomics data from the HMP2 cohort in IBDMDB. A majority voting consensus model was also generated using the outputs of the 7 individual trained models (Figure 9). This workflow demonstrates the advantage of using metatranscriptomic data to stratify patients. We expect that this technology could be improved in the future to enable the accurate identification of persister/dormant bacteria that may contain virulence factors.
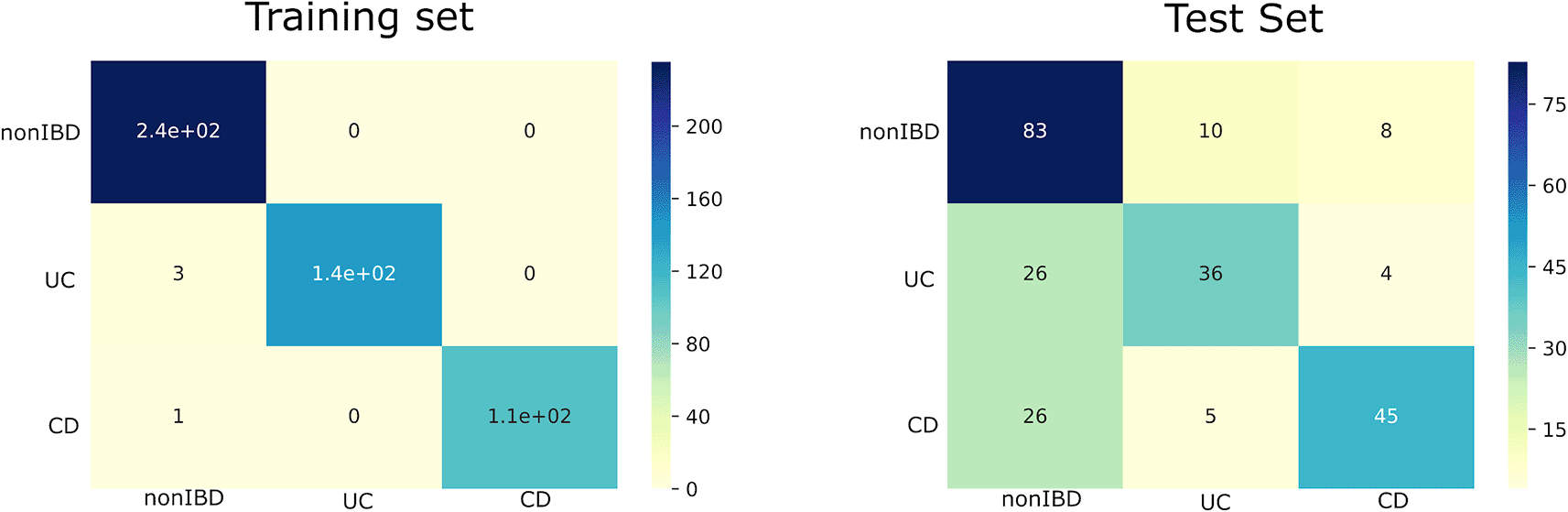
The training set (left) and test set (right). nonIBD: no Inflammatory Bowel Disease. UC: Ulcerative colitis. CD: Crohn's Disease.
This work describes our efforts to demonstrate the usefulness of using existing cloud-based platforms to not only perform analytical tasks related to identifying markers of microbial virulence, but also to use these platforms to store the relevant data and metadata. Specifically, we generated proof-of-concept computational workflows for data storage and indexing, metabolic functions, virulence factors, antimicrobial resistance, and mobile element detection in enterococci.
As metadata and derived metadata become more descriptive and comprehensive in the future, we expect that data indexing and storage will continue to play an important role in data mining activities. The subtask that involved comparing the complexity of compressed sequence data and metadata provided a foundation upon which to build and improve. We anticipate that the degree of metadata compression will be reduced as the metadata becomes more expansive and less repetitive overall. The increased metadata complexity could approach the complexity of the sequencing data in the future. The increased amounts of metadata would therefore help with both identifying relevant datasets by applying filtering criteria as well as with predicting additional related datasets. Overall, efforts for continued data indexing, together with the association of relevant and informative metadata are imperative to facilitate the future use and re-use of public datasets. In addition, future meta-analyses that both increase sample size and augment knowledge will benefit from enhancing existing minimum metadata reporting standards.
The ability to accurately identify metabolic functions associated with the disease or non-disease state from metagenomic data will be useful as sequencing-based assays continue to gain traction in the clinic. When sufficient datasets and metadata are used to train the machine learning model, this proof of concept shows that the presence of a disease state, and potentially even the identification of the specific disease state, can be possible. Similarly, identifying gene patterns that are specific to particular disease states and their contribution to disease progression would be invaluable as we seek to better understand the underlying mechanism(s) of disease.
We envision the computational identification of potential virulence factors to be useful not only in current data mining applications, but also to identify potential microbial factors that could be impacting human health once sequencing becomes more routine in the clinical setting. The results from a similar workflow could be used to inform the best treatment strategy for a given individual.
The computational AMR gene detection workflow that we implemented can be useful in clinical contexts where time from sample collection to prescribed treatment is precious. For example, rather than waiting for minimum inhibitory concentration (MIC) assay results to be performed and reported by the clinical lab, sequencing a sample and running an AMR-detection pipeline could inform the clinician which AMR genes are present in the sample and could nicely complement the MIC results.
The mobilome analysis will be used in cases where users are interested in the transmissibility of genomic material, particularly related to antimicrobial resistance. Association of resistance genes with transposons, plasmids, or other mobile elements can yield valuable information on the transmission of resistance through bacterial populations. This can help track the emergence and spread of antimicrobial resistance on particular genetic elements.
Overall, we found the NCBI hackathon for Microbial Virulence in the Cloud was extremely productive as measured by the various workflows, analyses, and results that were generated. These tools and results can be built upon by the scientific research community to improve existing methods and initiate important discussions about roadblocks and successes associated with our approach. For example, combining the various virulence detection methods could enable the rapid identification of emerging pathogens, while linking metabolism and signaling results with expression analyses could be used to identify novel indicator genes and therapeutic targets.
We are pleased with the publicly available workflows that were generated during this hackathon to build tools that can be applied to augment our understanding of microbial virulence and antimicrobial resistance. Potential next steps for this effort could include one or more of the following tasks: making the existing versions of pipelines and algorithms more robust, supporting the analysis of multiple input file types, applying and comparing the results from other algorithms, import workflow(s) into the NCBI Pathogen Detection Pipeline, dockerize tools and/or port them to external analysis platforms, improve data normalization methods, formalize the various database structure to facilitate data transfer across platforms, improve ease-of-use by developing web-based graphical user interfaces for a subset of tools, develop a database with the ability to take tab-delimited files containing metadata or other values and generate a table with standardized column names, and ensure the compatibility of tools, data structures, and containers across multiple cloud-based platforms.
Each of the workflows implemented during this hackathon greatly benefited from the availability of public data in the cloud. The analyses that were performed incorporated statistical analysis to minimize bias and quantify observed variability. Beginning each analysis from data and algorithms hosted in the cloud shows the relevance of cloud-based resources such as Google Big Query for storing, retrieving, and mining large datasets and repositories. This approach and the underlying cloud platform are both scalable in a way that should allow the support of other organisms and additional metadata. Throughout this process the extreme importance of accurate, descriptive, and available metadata became apparent. In addition, we found that augmenting the current metadata for each experiment by identifying features such as the presence of AMR or other virulence factors could improve downstream reuse and analysis. In summary, this hackathon quickly generated a number of publicly available workflows that can contribute to future research efforts related to AMR, metagenomics, and metatranscriptomics.
The data used for these projects were obtained from publicly accessible repositories, including NCBI, and are available in Table 1.
Pipelines and workflows available from:
• Virulence AMR Data Index Resource: https://github.com/NCBI-Hackathons/Virulence_AMR_Data_Index_Resource/
• Nasty Metagenomes: https://github.com/NCBI-Hackathons/Nasty_Metagenomes
• MetaClaMP-ML: https://github.com/NCBI-Hackathons/MetaClaMP-ML
• Virulence Factor Characterization: https://github.com/NCBI-Hackathons/Virulence_Factor_Characterization
• Enterococcus Mobile Elements: https://github.com/NCBI-Hackathons/Enterococcus_Mobile_Elements
• Enterococcus Mobile Elements: https://github.com/NCBI-Hackathons/Metatranscriptomics_Pilot
Archived pipelines and workflows as at time of publication:
• Virulence AMR Data Index Resource: https://doi.org/10.5281/zenodo.5227710 (DrCapt et al. 2021)
• Nasty Metagenomes: https://doi.org/10.5281/zenodo.5361759 (Mendes et al. 2021)
• MetaClaMP-ML: https://doi.org/10.5281/zenodo.5236567 (Elmassry et al. 2021)
• Virulence Factor Characterization: https://doi.org/10.5281/zenodo.5338778 (Payne et al. 2021)
• Enterococcus Mobile Elements: https://doi.org/10.5281/zenodo.5565736 (Ghtyson et al. 2021)
• Metatranscriptomics Pilot: https://doi.org/10.5281/zenodo.5227692 (Defne & Busby 2021)
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)