Keywords
bioinformatics, kinship, R, genealogy, SNPs, single nucleotide polymorphisms, relatedness
This article is included in the RPackage gateway.
This article is included in the Bioinformatics gateway.
bioinformatics, kinship, R, genealogy, SNPs, single nucleotide polymorphisms, relatedness
Inferring familial relationships between individuals using genetic data is a common practice in population genetics, medical genetics, and forensics. There are multiple approaches to estimating relatedness between samples, including genome-wide measures, such as those implemented in Plink1 or KING,2 and methods that rely on identity by descent (IBD) segment detection, such as GERMLINE,3 hap-IBD,4 and IBIS.5 Recent efforts focusing on benchmarking these methods6 have been aided by tools for simulating pedigrees and genome-wide SNP data.7 Analyzing results from genome-wide SNP-based kinship analysis or comparing analyses to simulated data for benchmarking have to this point required writing one-off analysis functions or utility scripts that are seldom distributed with robust documentation, test suites, or narrative examples of usage. There is a need in the field for a well-documented software package with a consistent design and API that contains functions to assist with downstream manipulation, benchmarking, and analysis of SNP-based kinship assessment methods. Here we present the skater package for SNP-based kinship analysis, testing, and evaluation with R.
The skater package provides an intuitive collection of analysis and utility functions for SNP-based kinship analysis. Functions in the package include tools for importing, parsing, and analyzing pedigree data, performing relationship degree inference, benchmarking relationship degree classification, and summarizing IBD segment data, described in full in the Use Cases section below. The package adheres to “tidy” data analysis principles, and builds upon the tools released under the tidyverse R ecosystem.8
The skater package is hosted in the Comprehensive R Archive Network (CRAN) which is the main repository for R packages: http://CRAN.R-project.org/package=skater. Users can install skater in R by executing the following code:
install.packages("skater")
Alternatively, the development version of skater is available on GitHub at https://github.com/signaturescience/skater. The development version may contain new features which are not yet available in the version hosted on CRAN. This version can be installed using the install_github() function in the devtools package:
install.packages("devtools")
devtools::install_github("signaturescience/skater", build_vignettes=TRUE)
When installing skater, other packages which skater depends on are automatically installed, including magritr, tibble, dplyr, tidyr, readr, purrr, kinship2, corrr, rlang, and others.
Minimal system requirements for installing and using skater include R (version 3.0.0 or higher) and several tidyverse packages8 that many R users will already have installed. Use cases are demonstrated in detail below. In summary, the skater package has functions for:
• Reading in various output files produced by commonly used tools in SNP-based kinship analysis
• Pedigree parsing, manpulation, and analysis
• Relationship degree inference
• Benchmarking and assessing relationship classification accuracy
• IBD segment analysis post-processing
A comprehensive reference for all the functions in the skater package is available at https://signaturescience.github.io/skater/.
The skater package provides a collection of analysis and utility functions for SNP-based kinship analysis, testing, and evaluation as an R package. Functions in the package include tools for working with pedigree data, performing relationship degree inference, assessing classification accuracy, and summarizing IBD segment data.
library(skater)
Pedigrees define familial relationships in a hierarchical structure. One of the common formats used by PLINK1 and other genetic analysis tools is the.fam file. A.fam file is a tabular format with one row per individual and columns for unique IDs of the mother, father, and the family unit. The package includes read_fam() to read files in this format:
famfile <- system.file("extdata", "3gens.fam", package="skater", mustWork=TRUE)
fam <- read_fam(famfile)
fam
## # A tibble: 64 x 6 ## fid id dadid momid sex affected ## <chr> <chr> <chr> <chr> <int> <int> ## 1 testped1 testped1_g1-b1-s1 0 0 1 1 ## 2 testped1 testped1_g1-b1-i1 0 0 2 1 ## 3 testped1 testped1_g2-b1-s1 0 0 1 1 ## 4 testped1 testped1_g2-b1-i1 testped1_g1-b1-s1 testped1_g1-b1-i1 2 1 ## 5 testped1 testped1_g2-b2-s1 0 0 1 1 ## 6 testped1 testped1_g2-b2-i1 testped1_g1-b1-s1 testped1_g1-b1-i1 2 1 ## 7 testped1 testped1_g3-b1-i1 testped1_g2-b1-s1 testped1_g2-b1-i1 2 1 ## 8 testped1 testped1_g3-b2-i1 testped1_g2-b2-s1 testped1_g2-b2-i1 1 1 ## 9 testped2 testped2_g1-b1-s1 0 0 2 1 ## 10 testped2 testped2_g1-b1-i1 0 0 1 1 ## #… with 54 more rows
Family structures imported from.fam formated files can then be translated to the pedigree structure used by the kinship2 package.9 The “fam” format may include multiple families, and the fam2ped() function will collapse them all into a tibble with one row per family:
peds <- fam2ped(fam) peds
## # A tibble: 8 x 3 ## fid data ped ## <chr> <list> <list> ## 1 testped1 <tibble [8 x 5]> <pedigree> ## 2 testped2 <tibble [8 x 5]> <pedigree> ## 3 testped3 <tibble [8 x 5]> <pedigree> ## 4 testped4 <tibble [8 x 5]> <pedigree> ## 5 testped5 <tibble [8 x 5]> <pedigree> ## 6 testped6 <tibble [8 x 5]> <pedigree> ## 7 testped7 <tibble [8 x 5]> <pedigree> ## 8 testped8 <tibble [8 x 5]> <pedigree>
In the example above, the resulting tibble is nested by family ID. The data column contains the individual family information, while the ped column contains the pedigree object for that family. Using standard tidyverse operations, the resulting tibble can be unnested for any particular family:
peds %>% dplyr::filter(fid=="testped1") %>% tidyr::unnest(cols=data)
## # A tibble: 8 x 7 ## fid id dadid momid sex affected ped ## <chr> <chr> <chr> <chr> <int> <dbl> <list> ## 1 testped1 testped1_g1-b1-s1 <NA> <NA> 1 1 <pedig~ ## 2 testped1 testped1_g1-b1-i1 <NA> <NA> 2 1 <pedig~ ## 3 testped1 testped1_g2-b1-s1 <NA> <NA> 1 1 <pedig~ ## 4 testped1 testped1_g2-b1-i1 testped1_g1-b1-s1 testped1_~ 2 1 <pedig~ ## 5 testped1 testped1_g2-b2-s1 <NA> <NA> 1 1 <pedig~ ## 6 testped1 testped1_g2-b2-i1 testped1_g1-b1-s1 testped1_~ 2 1 <pedig~ ## 7 testped1 testped1_g3-b1-i1 testped1_g2-b1-s1 testped1_~ 2 1 <pedig~ ## 8 testped1 testped1_g3-b2-i1 testped1_g2-b2-s1 testped1_~ 1 1 <pedig~
A single pedigree can also be inspected or visualized (standard base R plot arguments such as mar or cex can be used to adjust aesthetics):
peds$ped[[1]]
## Pedigree object with 8 subjects ## Bit size= 4
plot(peds$ped[[1]], mar=c(1,4,1,4), cex=.7)
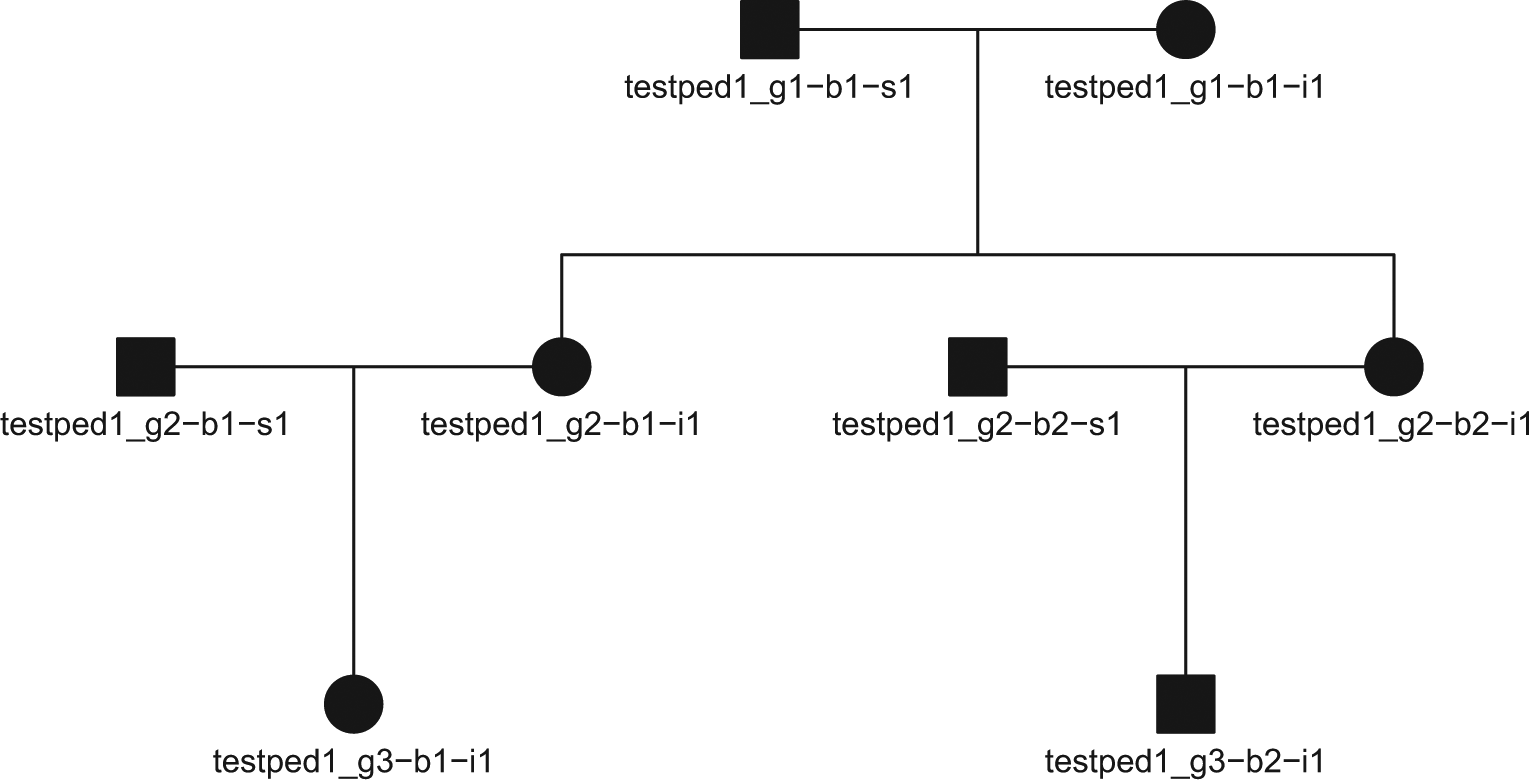
The plot_pedigree() function from skater will iterate over a list of pedigree objects, writing a multi-page PDF, with each page containing a pedigree from family:
plot_pedigree(peds$ped, file="3gens.ped.pdf")
The ped2kinpair() function takes a pedigree object and produces a pairwise list of relationships between all individuals in the data with the expected kinship coefficients for each pair.
The function can be run on a single family:
ped2kinpair(peds$ped[[1]])
## # A tibble: 36 x 3 ## id1 id2 k ## <chr> <chr> <dbl> ## 1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 ## 2 testped1_g1-b1-i1 testped1_g1-b1-s1 0 ## 3 testped1_g1-b1-s1 testped1_g2-b1-s1 0 ## 4 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 ## 5 testped1_g1-b1-s1 testped1_g2-b2-s1 0 ## 6 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 ## 7 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 ## 8 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 ## 9 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5 ## 10 testped1_g1-b1-i1 testped1_g2-b1-s1 0 ## # … with 26 more rows
This function can also be mapped over all families in the pedigree:
kinpairs <- peds %>% dplyr::mutate(pairs=purrr::map(ped, ped2kinpair)) %>% dplyr::select(fid, pairs) %>% tidyr::unnest(cols=pairs) kinpairs
## # A tibble: 288 x 4 ## fid id1 id2 k ## <chr> <chr> <chr> <dbl> ## 1 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 ## 2 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0 ## 3 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0 ## 4 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 ## 5 testped1 testped1_g1-b1-s1 testped1_g2-b2-s1 0 ## 6 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 ## 7 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 ## 8 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 ## 9 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5 ## 10 testped1 testped1_g1-b1-i1 testped1_g2-b1-s1 0 ## # … with 278 more rows
Note that this maps ped2kinpair() over all ped objects in the input tibble, and that relationships are not shown for between-family relationships.
The skater package includes functions to translate kinship coefficients to relationship degrees. The kinship coefficients could come from ped2kinpair() or other kinship estimation software.
The dibble() function creates a degree inference tibble, with degrees up to the specified max_degree (default=3), expected kinship coefficient, and lower (l) and upper (u) inference ranges as defined in Manichaikul et al.2 Degree 0 corresponds to self/identity/monozygotic twins, with an expected kinship coefficient of 0.5, with inference range >=0.354. Anything beyond the maximum degree resolution is considered unrelated (degree NA). Note also that while the theoretical upper boundary for the kinship coefficient is 0.5, the inference range for 0-degree (same person or identical twins) extends to 1 to allow for floating point arithmetic and stochastic effects resulting in kinship coefficients above 0.5.
dibble()
## # A tibble: 5 x 4 ## degree k l u ## <int> <dbl> <dbl> <dbl> ## 1 0 0.5 0.354 1 ## 2 1 0.25 0.177 0.354 ## 3 2 0.125 0.0884 0.177 ## 4 3 0.0625 0.0442 0.0884 ## 5 NA 0 -1 0.0442
The degree inference max_degree default is 3. Change this argument to allow more granular degree inference ranges:
dibble (max_degree = 5)
## # A tibble: 7 x 4 ## degree k l u ## <int> <dbl> <dbl> <dbl> ## 1 0 0.5 0.354 1 ## 2 1 0.25 0.177 0.354 ## 3 2 0.125 0.0884 0.177 ## 4 3 0.0625 0.0442 0.0884 ## 5 4 0.0312 0.0221 0.0442 ## 6 5 0.0156 0.0110 0.0221 ## 7 NA 0 -1 0.0110
Note that the distance between relationship degrees becomes smaller as the relationship degree becomes more distant. The dibble() function will emit a warning with max_degree >=10, and will stop with an error at >=12.
The kin2degree() function infers the relationship degree given a kinship coefficient and a max_degree up to which anything more distant is treated as unrelated. Example first degree relative:
kin2degree(.25, max_degree=3) ## [1] 1
Example 4th degree relative, but using the default max_degree resolution of 3:
kin2degree(.0312, max_degree=3) ## [1] NA
Example 4th degree relative, but increasing the degree resolution:
kin2degree(.0312, max_degree=5) ## [1] 4
The kin2degree() function is vectorized over values of k, so it can be used inside of a mutate on a tibble of kinship coefficients:
# Get two pairs from each type of relationship we have in kinpairs: kinpairs_subset <- kinpairs %>% dplyr::group_by(k) %>% dplyr::slice(1:2) kinpairs_subset
## # A tibble: 10 x 4 ## # Groups: k [5] ## fid id1 id2 k ## <chr> <chr> <chr> <dbl> ## 1 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0 ## 2 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0 ## 3 testped1 testped1_g3-b1-i1 testped1_g3-b2-i1 0.0625 ## 4 testped2 testped2_g3-b1-i1 testped2_g3-b2-i1 0.0625 ## 5 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 ## 6 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 ## 7 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 ## 8 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 ## 9 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 ## 10 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5
# Infer degree out to third degree relatives: kinpairs_subset %>% dplyr::mutate (degree=kin2degree(k, max_degree=3))
## # A tibble: 10 x 5 ## # Groups: k [5] ## fid id1 id2 k degree ## <chr> <chr> <chr> <dbl> <int> ## 1 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0 NA ## 2 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0 NA ## 3 testped1 testped1_g3-b1-i1 testped1_g3-b2-i1 0.0625 3 ## 4 testped2 testped2_g3-b1-i1 testped2_g3-b2-i1 0.0625 3 ## 5 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 2 ## 6 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 2 ## 7 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 1 ## 8 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 1 ## 9 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 0 ## 10 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5 0
Once estimated kinship is converted to degree, it may be of interest to compare the inferred degree to truth. When aggregated over many relationships and inferences, this approach can help benchmark performance of a particular kinship analysis method.
The skater package adapts a confusion_matrix() function from Clark10 to provide standard contingency table metrics (e.g. sensitivity, specificity, PPV, precision, recall, F1, etc.) with a new reciprocal RMSE (R-RMSE) metric. The confusion_matrix() function on its own outputs a list with four objects:
1. A tibble with calculated accuracy, lower and upper bounds, the guessing rate and p-value of the accuracy vs. the guessing rate.
2. A tibble with contingency table statistics calculated for each class. Details on the statistics calculated for each class can be reviewed on the help page for ?confusion_matrix.
3. A matrix with the contingency table object itself.
4. A vector with the reciprocal RMSE (R-RMSE). The R-RMSE represents an alternative to classification accuracy when benchmarking relationship degree estimation and is calculated using the formula in (1). Taking the reciprocal of the target and predicted degree results in larger penalties for more egregious misclassifications (e.g., classifying a first-degree relative pair as second degree) than misclassifications at more distant relationships (e.g., misclassifying a fourth-degree relative pair as fifth-degree). The +0.5 adjustment prevents division-by-zero when a 0th-degree (identical) relative pair is introduced.
To illustrate the usage, this example will start with the kinpairs data from above and randomly flip ~20% of the true relationship degrees:
# Function to randomly flip levels of a factor (at 20%, by default) randomflip <- function(x, p=.2) ifelse(runif(length(x))<p, sample(unique(x)), x)
# Infer degree (truth/target) using kin2degree, then randomly flip 20% of them set.seed(42) kinpairs_inferred <- kinpairs %>% dplyr::mutate(degree_truth=kin2degree(k, max_degree=3)) %>% dplyr::mutate(degree_truth=tidyr::replace_na(degree_truth, "unrelated")) %>% dplyr::mutate(degree_inferred=randomflip (degree_truth)) kinpairs_inferred
## # A tibble: 288 x 6 ## fid id1 id2 k degree_truth degree_inferred ## <chr> <chr> <chr> <dbl> <chr> <chr> ## 1 testped1 testped1_g1-b1-s1 testped1_g1-b1-s1 0.5 0 0 ## 2 testped1 testped1_g1-b1-i1 testped1_g1-b1-s1 0 unrelated unrelated ## 3 testped1 testped1_g1-b1-s1 testped1_g2-b1-s1 0 unrelated unrelated ## 4 testped1 testped1_g1-b1-s1 testped1_g2-b1-i1 0.25 1 1 ## 5 testped1 testped1_g1-b1-s1 testped1_g2-b2-s1 0 unrelated unrelated ## 6 testped1 testped1_g1-b1-s1 testped1_g2-b2-i1 0.25 1 1 ## 7 testped1 testped1_g1-b1-s1 testped1_g3-b1-i1 0.125 2 2 ## 8 testped1 testped1_g1-b1-s1 testped1_g3-b2-i1 0.125 2 1 ## 9 testped1 testped1_g1-b1-i1 testped1_g1-b1-i1 0.5 0 0 ## 10 testped1 testped1_g1-b1-i1 testped1_g2-b1-s1 0 unrelated unrelated ## #… with 278 more rows
Next, running the confusion_matrix() function will return all four objects noted above:
confusion_matrix(prediction = kinpairs_inferred$degree_inferred, target = kinpairs_inferred$degree_truth)
## $Accuracy ## # A tibble: 1 x 5 ## Accuracy ‘Accuracy LL‘ ‘Accuracy UL‘ ‘Accuracy Guessing‘ ‘Accuracy P-value‘ ## <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 0.812 0.763 0.856 0.333 1.09e-62 ## ## $Other ## # A tibble: 6 x 15 ## Class N ‘Sensitivity/Re~ ‘Specificity/TN~ ‘PPV/Precision‘ NPV ‘F1/Dice‘ ## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 0 64 0.75 0.964 0.857 0.931 0.8 ## 2 1 72 0.806 0.944 0.829 0.936 0.817 ## 3 2 48 0.833 0.967 0.833 0.967 0.833 ## 4 3 8 0.75 0.936 0.25 0.992 0.375 ## 5 unrelated 96 0.854 0.958 0.911 0.929 0.882 ## 6 Average 57.6 0.799 0.954 0.736 0.951 0.741 ## # … with 8 more variables: Prevalence <dbl>, Detection Rate <dbl>, ## # Detection Prevalence <dbl>, Balanced Accuracy <dbl>, FDR <dbl>, FOR <dbl>, ## # FPR/Fallout <dbl>, FNR <dbl> ## ## $Table ## Target ## Predicted 0 1 2 3 unrelated ## 0 48 4 2 1 1 ## 1 5 58 4 0 3 ## 2 0 3 40 1 4 ## 3 8 4 0 6 6 ## unrelated 3 3 2 0 82 ## ## $recip_rmse ## [1] 0.4665971
Standard tidyverse functions such as purrr::pluck() can be used to isolate just the contingency table:
confusion_matrix(prediction = kinpairs_inferred$degree_inferred,
target = kinpairs_inferred$degree_truth) %>%
purrr::pluck("Table")
## Target ## Predicted 0 1 2 3 unrelated ## 0 48 4 2 1 1 ## 1 5 58 4 0 3 ## 2 0 3 40 1 4 ## 3 8 4 0 6 6 ## unrelated 3 3 2 0 82
The confusion_matrix() function includes an argument to output in a tidy (longer=TRUE) format, and the example below illustrates how to spread contingency table statistics by class:
confusion_matrix(prediction = kinpairs_inferred$degree_inferred,
target = kinpairs_inferred$degree_truth,
longer = TRUE) %>%
purrr::pluck("Other") %>%
tidyr::spread (Class, Value) %>%
dplyr::relocate (Average, .after=dplyr::last_col()) %>%
dplyr::mutate_if(rlang::is_double, signif, 2)
## # A tibble: 14 x 7 ## Statistic ‘0‘ ‘1‘ ‘2‘ ‘3‘ unrelated Average ## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> ## 1 Balanced Accuracy 0.86 0.88 0.9 0.84 0.91 0.88 ## 2 Detection Prevalence 0.19 0.24 0.17 0.083 0.31 0.2 ## 3 Detection Rate 0.17 0.2 0.14 0.021 0.28 0.16 ## 4 F1/Dice 0.8 0.82 0.83 0.38 0.88 0.74 ## 5 FDR 0.14 0.17 0.17 0.75 0.089 0.26 ## 6 FNR 0.25 0.19 0.17 0.25 0.15 0.2 ## 7 FOR 0.069 0.064 0.033 0.0076 0.071 0.049 ## 8 FPR/Fallout 0.036 0.056 0.033 0.064 0.042 0.046 ## 9 N 64 72 48 8 96 58 ## 10 NPV 0.93 0.94 0.97 0.99 0.93 0.95 ## 11 PPV/Precision 0.86 0.83 0.83 0.25 0.91 0.74 ## 12 Prevalence 0.22 0.25 0.17 0.028 0.33 0.2 ## 13 Sensitivity/Recall/TPR 0.75 0.81 0.83 0.75 0.85 0.8 ## 14 Specificity/TNR 0.96 0.94 0.97 0.94 0.96 0.95
Tools such as hap-IBD,4 and IBIS5 detect shared IBD segments between individuals. The skater package includes functionality to take those IBD segments, compute shared genomic centimorgan (cM) length, and converts that shared cM to a kinship coefficient. In addition to inferred segments, these functions can estimate “truth” kinship from simulated IBD segments.7 The read_ibd() function reads pairwise IBD segments from IBD inference tools and from simulated IBD segments. The read_map() function reads in genetic map in a standard format which is required to translate the total centimorgans shared IBD to a kinship coefficient using the ibd2kin() function. See ?read_ibd and ?read_map for additional details on expected format.
The read_ibd() function reads in the pairwise IBD segment format. Input to this function can either be inferred IBD segments from hap-IBD (source="hapibd") or simulated segments (source="pedsim"). The first example below uses data in the hap-ibd output format:
hapibd_filepath <- system.file("extdata", "GBR.sim.ibd.gz",
package="skater")
hapibd_seg <- read_ibd(hapibd_filepath, source = "hapibd")
hapibd_seg
## # A tibble: 3,954 x 6 ## id1 id2 chr start end length ## <chr> <chr> <dbl> <dbl> <dbl> <dbl> ## 1 testped1_g1-b1-s1 testped1_g3-b1-i1 1 197661576 234863602 47.1 ## 2 testped1_g2-b2-i1 testped1_g3-b1-i1 1 197661576 231017545 39.8 ## 3 testped1_g3-b1-i1 testped1_g3-b2-i1 1 197661576 212799139 20.3 ## 4 testped3_g1-b1-s1 testped3_g3-b2-i1 1 2352146 10862397 17.7 ## 5 testped3_g2-b2-i1 testped3_g3-b2-i1 1 2352146 10862397 17.7 ## 6 testped1_g1-b1-s1 testped1_g2-b1-i1 1 3328659 64123868 86.4 ## 7 testped1_g1-b1-s1 testped1_g3-b1-i1 1 3328659 33476811 51.2 ## 8 testped1_g2-b2-s1 testped1_g3-b2-i1 1 5003504 32315147 45.9 ## 9 testped2_g1-b1-i1 testped2_g3-b1-i1 1 240810528 248578622 15.9 ## 10 testped2_g1-b1-i1 testped2_g2-b2-i1 1 241186056 249170711 15.5 ## # … with 3,944 more rows
In order to translate the shared genomic cM length to a kinship coefficient, a genetic map must first be read in with read_map(). Software for IBD segment inference and simulation requires a genetic map. The map loaded for kinship estimation should be the same one used for creating the shared IBD segment output. The example below uses a minimal genetic map that ships with skater:
gmap_filepath <- system.file("extdata", "sexspec-avg-min.plink.map",
package="skater")
gmap <- read_map(gmap_filepath)
gmap
## # A tibble: 28,726 x 3 ## chr value bp ## <dbl> <dbl> <dbl> ## 1 1 0 752721 ## 2 1 0.0292 1066029 ## 3 1 0.0829 1099342 ## 4 1 0.157 1106473 ## 5 1 0.246 1152631 ## 6 1 0.294 1314015 ## 7 1 0.469 1510801 ## 8 1 0.991 1612540 ## 9 1 1.12 1892325 ## 10 1 1.41 1916587 ## # … with 28,716 more rows
The ibd2kin() function takes the segments and map file and outputs a tibble with one row per pair of individuals and columns for individual 1 ID, individual 2 ID, and the kinship coefficient for the pair:
ibd_dat <- ibd2kin(.ibd_data=hapibd_seg, .map=gmap) ibd_dat
## # A tibble: 196 x 3 ## id1 id2 kinship ## <chr> <chr> <dbl> ## 1 testped1_g1-b1-i1 testped1_g1-b1-s1 0.000316 ## 2 testped1_g1-b1-i1 testped1_g2-b1-i1 0.261 ## 3 testped1_g1-b1-i1 testped1_g2-b2-i1 0.263 ## 4 testped1_g1-b1-i1 testped1_g2-b2-s1 0.000150 ## 5 testped1_g1-b1-i1 testped1_g3-b1-i1 0.145 ## 6 testped1_g1-b1-i1 testped1_g3-b2-i1 0.133 ## 7 testped1_g1-b1-i1 testped2_g1-b1-i1 0.000165 ## 8 testped1_g1-b1-i1 testped2_g1-b1-s1 0.000323 ## 9 testped1_g1-b1-i1 testped2_g2-b1-i1 0.000499 ## 10 testped1_g1-b1-i1 testped2_g2-b1-s1 0.000318 ## # … with 186 more rows
The skater R package provides a robust software package for data import, manipulation, and analysis tasks typically encountered when working with SNP-based kinship analysis tools. All package functions are internally documented with examples, and the package contains a vignette demonstrating usage, inputs, outputs, and interpretation of all key functions. The package contains internal tests that are automatically run with continuous integration via GitHub Actions whenever the package code is updated. The skater package is permissively licensed (MIT) and is easily extensible to accommodate outputs from new genome-wide relatedness and IBD segment methods as they become available.
1. Software available from: http://CRAN.R-project.org/package=skater.
2. Source code available from: https://github.com/signaturescience/skater.
3. Archived source code at time of publication: https://doi.org/10.5281/zenodo.5761996.11
4. Software license: MIT License.
This work was supported in part by award 2019-DU-BX-0046 (Dense DNA Data for Enhanced Missing Persons Identification) to B.B., awarded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice and by internal funds from the Center for Human Identification. The opinions, findings, and conclusions or recommendations expressed are those of the authors and do not necessarily reflect those of the U.S. Department of Justice.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)