Keywords
active contours, banding information, clustering, convex-concave, di-centric, geometric features, learning architectures, overlapping, thresholding, touching
This article is included in the Bioinformatics gateway.
active contours, banding information, clustering, convex-concave, di-centric, geometric features, learning architectures, overlapping, thresholding, touching
The structural and numerical abnormalities in the chromosomes cause genetic disorders. Therefore, chromosome analysis is a key method in diagnosing genetic disorders. Standard method of chromosome analysis is known as karyotyping. Conventional karyotyping classifies a metaphase image into 24 classes manually.1,2 Karyotyping requires expertise which may subject to inter-observer variability and intra-observer variability. In addition, it is a time-consuming and tedious process which may result in fatigue errors.3
To address the aforementioned limitations of conventional manual karyotyping, automated human chromosome segmentation and feature extraction was introduced. Automated processes are repeatable because of the computer-based performance, hence no observer variabilities occur. Expertise is not required to handle chromosome segmentation and feature extraction algorithms. Moreover, these algorithms address the time in-efficiency of tedious manual process.
The automated karyotyping process consists of four main steps; image pre-processing, chromosome segmentation, chromosome feature extraction and clinical evaluation.4–6 This review paper extensively discusses two main steps among them; chromosome segmentation and chromosome feature extraction. Figure 1 illustrates the scope of the paper. Section II presents a brief background of karyotyping, including the methods of staining, methods of visualization and publicly available datasets of metaphase chromosome images for research purposes. Sections III and IV provide a comprehensive explanation of the approaches towards automated chromosome segmentation and chromosome feature extraction respectively. Finally, section V provides suggestions for the future improvement of automated human chromosome segmentation and feature extraction.

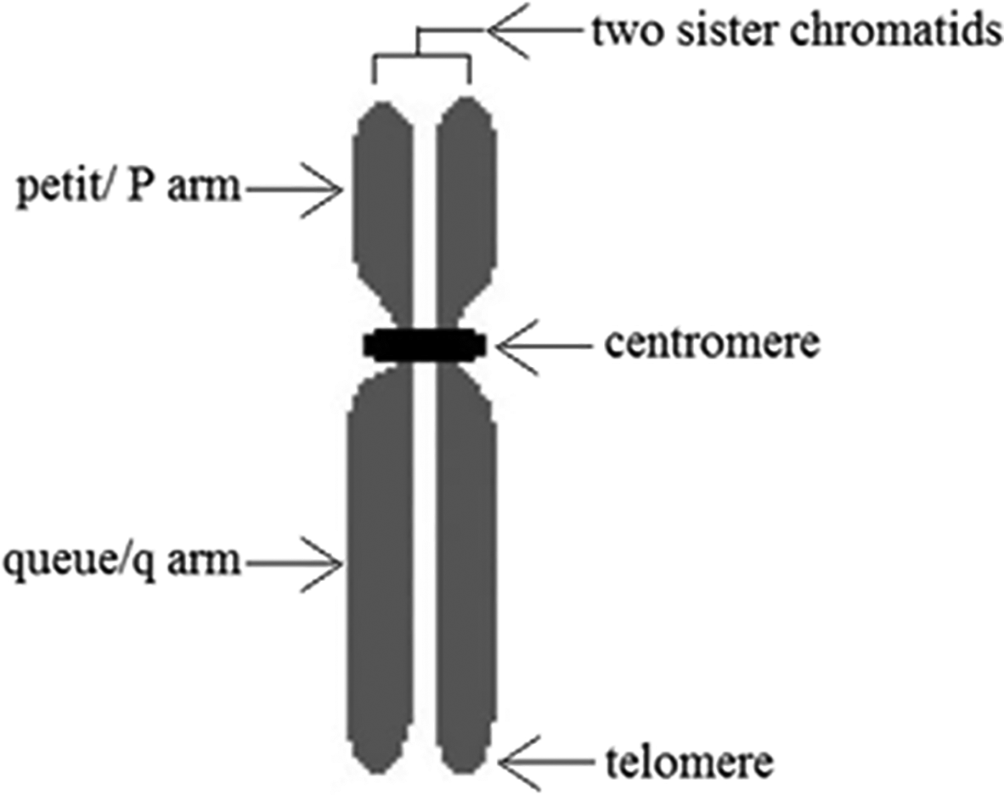
Each chromosome in cell nuclei contains two sister chromatids which separate from each other in cell division. During metaphase, two chromatids attach to each other only at centromere, as shown in Figure 2.7 These chromatids contain unique sub-structures of genes which can be visualized by staining. Condensed areas of the chromosomes appear in dark bands while less condensed areas appear in light bands. Therefore, each chromatid produces a unique banding pattern after staining according to its sub-structures.7–9 Different types of staining such as G-banding,10 R-banding, C-banding,11 Q-banding,10,12 NOR banding and T-banding13 are available for chromosome staining. 4,6-diamidino-2-phenylindole (DAPI) is a fluorescent stain which is commonly used for chromosome staining. The reason for this common use is, the possibility to avoid the need of preparation of multiple duplicate cell samples.14
After the staining of chromosomes, a visible photographic representation of them is produced by using different imaging techniques such as light microscopy, fluorescent microscopy, electron microscopy and coherent x-ray diffraction imaging.15,16 However, producing original data might be difficult and inefficient in some research work. As a solution, publicly available databases of visible photographic representations of the stained chromosomes can be used. Edinburgh, Philadelphia,17 Copenhagen,18 BioImlab,19 Lisbon-K1 (LK1)20 and M-FISH21 are few examples for publicly available databases which can be accessed by the researchers.
The visualized stained chromosomes are paired and ordered manually to produce the karyotype during the conventional karyotyping.22 This is a time-consuming, tedious process23 and requires expertise to produce the karyotype accurately. In addition, fatigue errors and observer variabilities may occur due to the repetitive process. To address these limitations of inefficient manual karyotyping, semi-automated karyotyping was introduced in late 1970s.17,24 Later, the development of the computer processors and digital image processing paved the path towards fully-automated human chromosome segmentation and feature extraction.25 However, there are multiple challenges in automating this process as explained in the next sub-section.
The banding patterns which appear in the chromosome images depend on the method of staining. Therefore, developing general algorithms applicable to any type of staining is challenging. The majority of the chromosome image segmentation techniques are limited to the images produced by one staining method. It limits the application of these chromosome segmentation algorithms in practical scenario.7–9
High resolution images are required to improve the accuracy of the process of segmentation and feature extraction. However, using the visualization techniques which produce high resolution images may damage the chromosomes. Despite the availability of different advanced imaging techniques, highly sensitive and specific methods are required to provide high resolution chromosome images with insignificant damage to the chromosomes. Developing imaging techniques which fulfil the aforementioned requirement, remains a challenge.16,26,27
Chromosome images are distorted with artifacts such as staining and cell debris.28,29 Therefore, additional steps are required to filter the artifacts for accurate chromosome segmentation and feature extraction. However, over-filtering may lead to loss of information. Therefore, filtering the chromosome images while preserving the important information of the chromosomes poses a challenge.
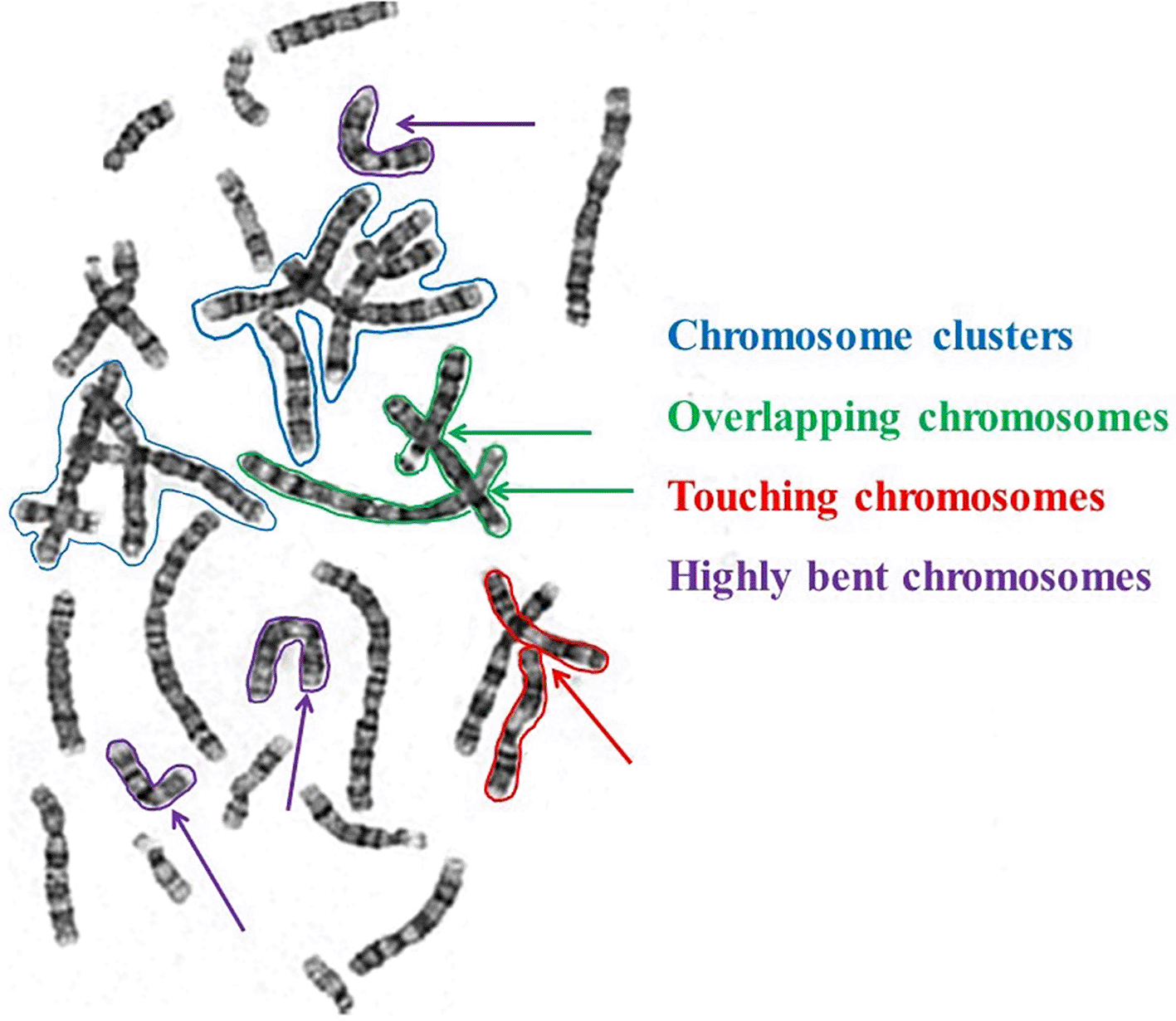
In addition, presence of overlapping, touching and bent chromosomes,28 morphological variations, and presence of partial sister-chromatid separation or inter-phase cells30 raise a challenging nature in automating the chromosome segmentation and feature extraction. Figure 3 illustrates the appearance of overlapping chromosome clusters, overlapping and touching chromosome clusters, and highly bent chromosomes in the image.
Chromosome segmentation is the process of extracting the chromosomes from the image. This section extensively discusses chromosome segmentation algorithms under four sub-categories; thresholding, clustering, active contours and convex-concave points-based methods.
Thresholding relies on the intensity differences between the chromosomes and the background. Thresholding works for the chromosome images because chromosomes tend to appear in bright intensities compared to the background of the image.31–33
Common global thresholding techniques which have been used to segment the chromosome images include Otsu’s thresholding and Kapur’s thresholding. Otsu’s thresholding extracts the chromosomes from the image by minimizing the intra-class variance of the pixels within each cluster.34–36 Kapur’s global thresholding divides a chromosome image into foreground and background based on the assumption that the chromosomes and the background of the image carry two different probability distributions. The optimal threshold level for Kapur’s thresholding is obtained by maximizing the total entropy.35 However, global thresholding37 often fails due to intensity in-homogeneity caused by the staining and lighter intensities of smaller chromosomes compared to larger chromosomes. Moreover, global thresholding can detect the chromosome clusters but fails in disentangling the cluster.
Consequently, local thresholding methods were introduced to segment the chromosome images.31,32,38,39 Adaptive local thresholding provides better results comparatively.35 However, simple thresholding-based techniques create holes inside the chromosomes when pixels inside the chromosomes are lighter than the surrounding pixels and, the boundaries might be cropped due to intensity fading.35 Sensitivity of the thresholded image to the quantization error of the digital image is another issue in thresholding-based chromosome segmentation.40
In addition, simple thresholding-based techniques do not work when overlapping chromosomes present in the image because two overlapping chromosomes share a significant area in the histogram.17,41,42 When touching chromosomes present in the image, they are detected as a single chromosome. Therefore, the applicability of the thresholding-based segmentation methods is limited. Consequently, when overlapping and touching chromosomes present in the images, human intervention is needed to segregate them.43–45
A detailed review on thresholding-based chromosome segmentation is presented below.
Poletti et al.46 presented a review on thresholding strategies applied for the chromosome image segmentation. According to this review paper, thresholding is the first step in majority of the chromosome segmentation algorithms. Poletti et al. implemented 11 approaches which include thresholding at least in one step. The implemented algorithms are as follows; Otsu’s thresholding, Kapur’s thresholding, improved Sobel with genetic algorithm, K-means clustering on algebraic moments, Fuzzy C-mean clustering, multi-stage adaptive thresholding, local re-thresholding, adaptive thresholding, region-based level sets, multi-thresholding with Particle Swarm and Nelder–Mead optimization, and multi-thresholding with Particle Swarm and expectation-maximization Optimization. The performance of these algorithms were compared with each other using Q-band images. Overall best performance was achieved for adaptive local thresholding and region-based level sets.
Yilmaz et al.12 introduced a method for accurate segmentation of touching and overlapping chromosomes. To obtain the smooth and circular shape of the chromosomes, morphological closing was applied to the binary image several times. Adaptive local thresholding proposed by Grisan et al.38 was used to filter the debris. However, a gray level threshold map along with a fixed size Gaussian kernel was used instead of the method of dividing the image into squares as proposed by Grisan et al. The method based on watershed transform proposed by Karvelis et al.47 was used to reconstruct a distance-transformed image. Clusters were identified by considering the average area of 46 chromosomes, convex hull, average thickness of the chromosomes and thickness along the skeleton. Nicholas Howe’s implementation48 was used for skeletonization. Concave points and gray-scale geodesic path were considered when separating the touching chromosomes. Overlapping chromosomes were separated by generating candidate combinations. This study achieved 97.8% accuracy with 6,678 chromosomes which was superior to the existing literature.
Grisan et al.38 introduced a local adaptive thresholding for Q-band chromosome image segmentation. First, the image was divided into tessellation of squares of fixed dimension which include 100 pixels in each. Next, Otsu’s thresholding was applied to each square. After detecting chromosome clusters, medial axis transform, analysis of accumulation points, single chromosome likelihood measure, generation of candidate splits and, generation and exploration of the hypotheses tree were used to disentangle the chromosomes. The dataset used by Grisan et al. includes 6,683 chromosomes. Overall accuracy for Q-band image segmentation using this algorithm was 94%. This algorithm achieved 90% accuracy for both touching chromosomes and overlapping chromosomes.
Sugapriya et al.31 introduced a method to extract chromosomes from G-band metaphase images. This algorithm consists of three basic steps; local thresholding, region growing and, edge detection using gradient and Laplacian. The method of calculating the threshold value for local thresholding was as follows. First, the image was divided into sub-images. Next, the global threshold of each sub-image and largest element of the sub image was computed. The product of these two computed values was taken as the local threshold. However, quantitative analysis of the results on accuracy was not presented in the paper.
Di-centric chromosomes can be used to evaluate the radiation exposure. In the light of the above, a modified watershed-based method to segment the overlapped chromosomes was introduced by Li et al.49 First, the local thresholding was applied and foreground pixels were identified. Next, watershed transform was used to separate the chromosomes. Then, inseparable clusters and debris were filtered by using morphological decision tree. However, this was not a promising solution because the algorithm failed to segment the chromosomes accurately when considerable sister chromatid separation and non-uniform staining presented.
Stanley et al.39 introduced a homologous matching algorithm to segment chromosome images. First, the image was splitted into two using global thresholding. Next, local thresholding was applied to thresholded image for object separation. Finally, each chromosome was detected using five features; size, centromeric index, banding information, profiles along the medial axis and weighted density distribution.
Ji et al.32 proposed a two-step method to segment chromosomes. In the first step, chromosome image was thresholded by using the main peak of the gray level histogram as threshold. Conservative model was utilized to avoid over segmentation. After this initial segmentation using global thresholding, another step of re-thresholding was performed to classify touching and overlapping chromosomes. In order to do that, the chromosomes were classified into classes depending on size and shape. Single chromosomes were detected by their smaller area. Next, touching chromosomes were detected using concavity and density information. Overlapping chromosomes were separated using three assumptions; at least one node is included in the overlapping cluster, four’cut points’ present around the node, and mean density around the node is higher.
Altinsoy et al.50 used U-net based Convolution Neural Network (CNN) for chromosome segmentation. First, the image was filtered using a thresholding-based filtering technique. Next, the irrelevant objects were removed by considering the boundary of the image and, shape and size of the objects. Then, watershed transform, adaptive filtering and successive iteration were used to separate the touching chromosomes. To improve the accuracy of classification of single objects and chromosome clusters, 13 parameters were employed. Few of these parameters were not discussed in the previous literature towards classification. The parameters include object size, limited object size, average object size, maximum single chromosome size, ellipse, object pixel intensity, global pixel intensity, skeleton, thickness throughout the skeleton, skeleton size, average thickness, number of branch points, and number of end points. In this approach, cutting points and cutting paths of the clusters were identified using concavity properties and geodesic distance transform, Euclidean distance and, shape of the node. Another merit of the algorithm was the capability in handling multiple overlaps. This algorithm achieved 96.97% accuracy for 40 distorted chromosome images proving that neural networks work better for distorted images. However, applicability of this method might be limited due to the small dataset. Therefore, further research using a large dataset is suggested to verify the conclusion made.
Clustering is one of the simplest and fastest methods of image segmentation.
Clustering-based algorithms can be divided into two as hierarchical algorithms and partitional algorithms. Both types of clustering-based algorithms work with intensity in-homogeneity and, have provided promising results in chromosome segmentation compared to simple thresholding-based algorithms.40 Hierarchical clustering algorithms provide higher accuracy compared to partitional clustering algorithms. However, the use of hierarchical clustering algorithms is limited due to the computational complexity. K-means and Fuzzy C-Mean (FCM) clustering algorithms, which are easy to implement and are low in computational complexity, have been used in research work to a greater extent.51,52 However, Fuzzy clustering performs better in chromosome segmentation compared to K-means clustering. Accuracy of the clustering-based algorithms can be improved by incorporating watershed transform.53
Clustering-based algorithms are relatively less time and computer complex. However, these algorithms require manual intervention in selecting the seed point to initiate. A slight mistake in selecting the seed point might lead to inaccurate segmentation. In addition, the output is highly sensitive to the number of clusters which is a pre-set parameter, hence expertise is required. Moreover, clustering-based algorithms are highly sensitive to outliers. Therefore, clustering-based algorithms often result in inaccurate chromosome segmentation, specially when stain debris and cell debris present in the image.
This section continues the discussion on clustering-based chromosome segmentation algorithms using the approaches presented in the literature.
Sayed and Hassanien29 proposed two methods to perform accurate chromosome segmentation when inter-phase cells present in the metaphase chromosome images. The first method is based on FCM and Grey Wolf Optimization. The second method is based on hybrid particle swarm optimization54,55 and K-Means clustering. Generic algorithms were used to select the best fit cluster in this algorithm. The experimental results had shown 94% and 95% accuracy for the methods respectively.
Munot et al.40 introduced a Random Walker Algorithm (RWA)-based clustering for accurate chromosome segmentation despite the scale and, the presence of chromosome clusters and highly bent chromosomes. Furthermore, this method overcomes the limitation of working only with grayscale images. This algorithm was tested using a publicly available M-FISH database. Its output was compared with the outputs of conventional Otsu’s thresholding and watershed transform. Main fact that limited the application of this algorithm was the need of precise seed point selection to avoid inaccurate segmentation.
Shen et al.53 introduced a novel algorithm to segment di-centric chromosomes based on k-means clustering and the watershed transform. First, minimum circumscribed rectangle was used to identify the curvatures in a single chromosome area. When the ratio between chromosome area to the smallest rectangle was close to 1, the chromosome was straight. For extended type of chromosomes, the long side of the circumscribed rectangle was parallel to the centerline. To extract the centerline of the severely bent chromosomes, the method used by Zhang and Suen was modified.56 This modification improved the accuracy by removing the small holes created inside the chromosomes and spurious branches at the end of the sister chromatids.
Manohar et al.57 compared the performance of FCM and watershed transform in chromosome segmentation using publicly available M-FISH database. The overall accuracy was 94% and 92% for FCM and watershed transform respectively. However, the dataset of this approach included only 20 images which limited the reliability of the output.
Kanimozhi et al. introduced a novel techniques to segment the chromosome images using quasi-Newton-based K-means clustering which worked for M-FISH images. M-FISH image characterization was performed using expectation-maximization-based hierarchical Bayes model. In this approach, misclassifications were avoided by using contextual-based classification and region merging. The researchers modified the activation function of sigmoid and softmax layer of the AlexNet for the optimum classification of autosomal chromosomes and sex chromosomes. This algorithm outperformed the existing literature with a 6.96% higher accuracy.
Li et al. addressed the low accuracy of FCM in M-FISH images by incorporating spatial and spectral FCM. It provided the advantage of noise reduction by exploiting information from neighboring pixels. In addition, it had the ability to incorporate pixel information across different channels simultaneously. The results obtained from the proposed method were compared with other FCM-based methods using accuracy and false-positive detection. A higher segmentation accuracy and a lower false-positive ratio were obtained for this algorithm, proving that both spatial and spectral data can enhance the performance of FCM-based methods for chromosomal segmentation.58
Active contours work with concavities of the chromosomes and create closed curves around the chromosomes. Therefore, active contours are comparatively accurate in segmenting the chromosomes compared to other categories given in this review paper. Active contours have the ability to resolve touching chromosomes; however, considerable improvement in the performance has not been achieved for overlapping chromosomes.59
Active contours can generate closed parametric curves around the chromosomes. However, they fail to extract chromosomes from the image when highly concave boundaries appear. Therefore, poor convergence property of active contours results in inaccurate segmentation specially when highly bent chromosomes present in the image.
Incorporating an external force model such as Gradient Vector Flow (GVF), resolves the aforementioned limitation of active contours. Discrete Cosine Transform (DCT) embedded into GVF is capable in providing a better description of energy levels of chromosomes in the image. Hence, DCT embedded GVF incorporated active contours output accurate segmentation.60
However, active contours suffer from the need of optimum parameter setting. In addition, automated initialization is difficult when artifacts present in the image. Level sets is a promising solution in the cost of time efficiency when minimal artifacts present in the image.
The rest of this section presents a review on related work on active contour-based chromosome segmentation.
Arora1 introduced a method to disentangle the overlapping and touching chromosomes irrespective of the staining method. Objects in the image of metaspread chromosomes were extracted using a region-based active contour model incorporating level sets used by Minaee et al.43 This algorithm worked properly even when weak boundaries and intensity inhomogeneity presented. However, region-based level sets consume higher execution time and might be inaccurate when artifacts present in the image.12 After segmentation, objects were classified as single chromosomes, chromosome clusters or artifacts considering the area, circularity and length. To separate touching chromosomes from overlapping chromosomes, gradient paths were considered. Over-segmentation was avoided by using region merging. This algorithm outputs 96% and 81% of accuracy for touching chromosomes and overlapping chromosomes respectively.
Hu et al.42 proposed an algorithm to address the requirement of human intervention for segmentation when partially overlapping chromosomes presented in the image. It was developed using U-net artificial neural network based on active contours. In fact, the model was improved in terms of computational time and storage by reducing the layers and blocks of the network to a minimum. This model was trained using 64% of the data, validated using 16% of the data, and tested on the remaining 20% of the data. The results showed intersection over union accuracy, 94.7% and 88-94% for overlapped regions and non-overlapped regions respectively.
Hu et al.61 proposed snake active contour-based CNN for chromosome segmentation. Edge-based snake active contour was used for manual annotation in the training process. The seed points were manually provided near to the outline of the chromosomes during the training process. Advantages of using edge-based snake model for training include avoiding over-segmentation of chromosome boundaries and low computer complexity. The dataset of this approach includes 4,184 chromosomes. However, under-fitting and over-fitting were observed due to low resolution of the image, blurry boundaries and natural defects of the snake models.
Human metaphase chromosomes can be segmented using curvature function which is built upon the concept of convexity and concavity.34,62 Centromere is the global minimum point of the image intensity profile63 which is presented in Figure 4. Concave-convex-based chromosome segmentation algorithms are capable in extracting both touching and overlapping chromosomes. They work with non-rigid chromosome boundaries. However, they often fail when chromosome clusters and highly-bent chromosomes appear in the image. Nevertheless, incorporating convex-concavity theorem to other segmentation categories results in comparatively accurate segmentation.1,28

Summary of the selected methods which use the theory of convex-concave points is given in Table 1. Majority of the approaches uses concave-convex points in the algorithms to improve the performance.
| Publication | Method |
|---|---|
| Madian et al.63 | Concave function |
| Madian et al.2 | Curvature function |
| Madian et al.34 | Incremental curvature function, angle of inclination, concave property of a region, separation lines, min set |
| Karvelis et al47 | Watershed transform based algorithm |
| Yuan et al.64 | Multi-scale corner detection method based on Hua’s method on Otsu’s thresholded image |
In 2018, a new method was implemented by Maidan et al.65 to address the limitation of segmenting either touching chromosomes or overlapping chromosomes using Giemsa banding images. First, the stained microscopic images were binarized. Then, the holes inside the chromosomes were filled using morphological operations to address one limitation of simple thresholding-based segmentation, i.e. creating holes inside the chromosomes when inner pixels appear in light intensities. Then, the contours of the chromosomes were approximated using deformation function. Interesting points, i.e. concave points (convex point is optional), were estimated using the curvature function. Finally, touching and overlapping chromosomes were segregated with 97.09% and 95% accuracy respectively based on interesting points and neighborhood connectivity property.
Table 2 presents a summary on the discussed human metaphase chromosome segmentation categories in this review paper including their strengths and limitations.
Incorporating learning architectures to automate the process of chromosome segmentation, is the current trend. Besides, reducing the blocks and the layers in order to minimize the computer complexity, consequently maximize the time efficiency and storage efficiency is required. Most of the segmentation algorithms are limited to one type of staining method which is a main limitation. In addition, it is important to consider the inter-phase cells present in the image during chromosome segmentation.66–68
Accurate centromere detection is needed in many applications such as determining the radiation exposure of an individual. Centerline (medial axis) detection aids in detecting the centromere of the chromosome and consequently, the abnormalities in it. Accurate chromosome segmentation is required for precise chromosome feature extraction.69 There are two major approaches in chromosome feature extraction based on banding information and based on geometric features. Two sub-sections presented hereafter in this section discuss the approaches under the aforementioned categories.
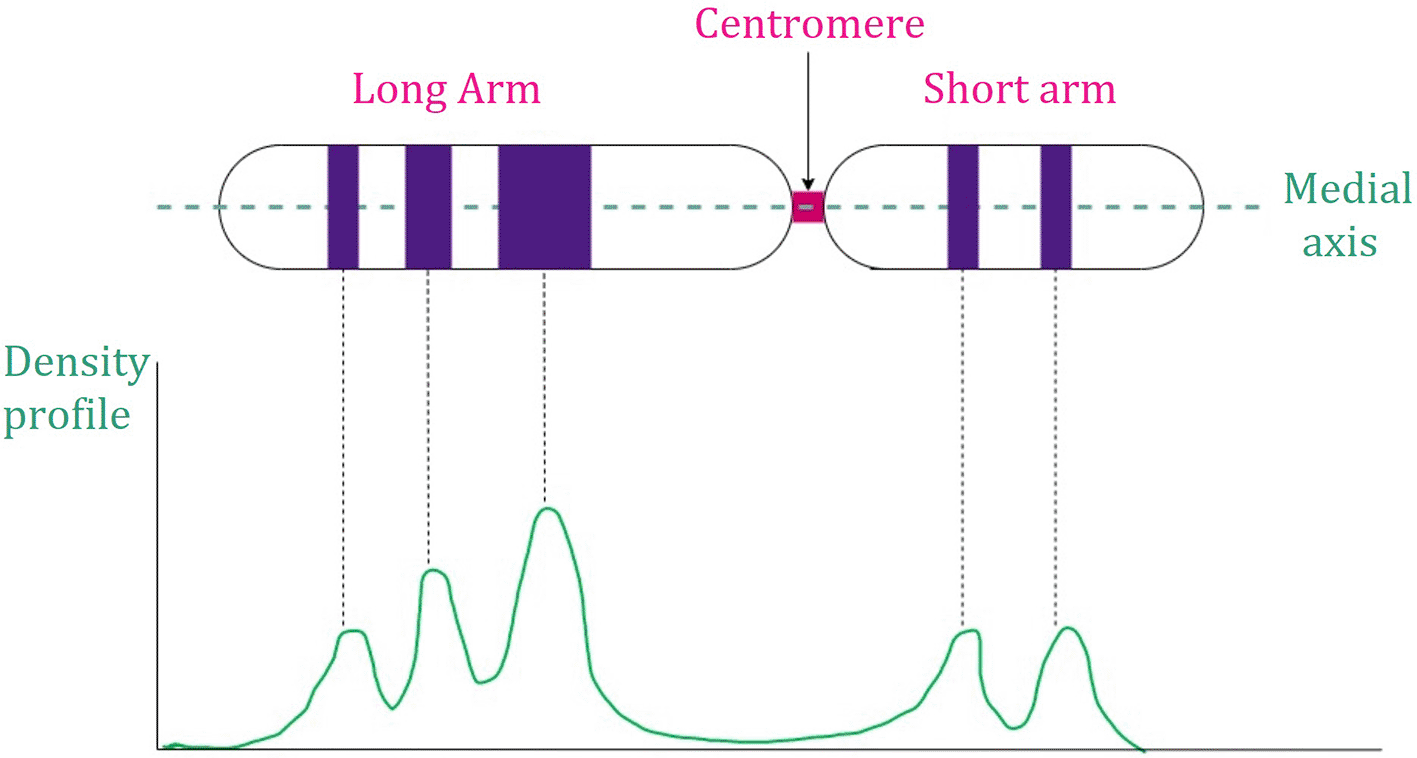
Banding information of the chromosomes depends on the banding pattern visualized by the staining methods. It can be presented by a density profile as illustrated in Figure 5. These methods are restricted to a particular type of staining. Consequently, the use of these methods is limited.69
Wang et al.70 proposed image enhancement algorithm based on a family of differential wavelet transforms to improve the contrast of the banding pattern of the chromosome images which is distorted due to cell culturing, staining and imaging conditions. However, the objective of this approach was to extract the geometric features including edges. Multi-scale point-wise product was used to improve the accuracy as geometric features correlate in different scales. This approach provided better classification results against three convolution-based methods; adaptive contrast stretch, adaptive contrast enhancement and contrast gain transform. In addition, time efficiency was improved.71
Errington et al.72 proved that considering only the banding pattern results in less accuracy. This approach was tested using Copenhagen, Edinburgh and Philadelphia databases. Higher accuracy was achieved when normalized length and centromeric index were considered together with the banding pattern.
The majority of the chromosome classification algorithms work on straight chromosomes to improve the classification accuracy.73,74 Sharma et al.75 proposed an algorithm based on deep CNN and Residual Neural Networks (ResNet) assuming that the chromosomes were segmented and straightened. Banding patterns appeared in dark and light intensities in the chromosome images were identified using recurrent neural network based on sequence learning. This algorithm was tested using 5,256 chromosomes and compared with traditional deep CNN, ResNet-50, CRNN and attention-based sequence model. The proposed algorithm (Res-CRANN) outperformed the other modules in terms of accuracy and robustness.75
Jindal et al.76 used the information on banding patterns to classify the chromosomes using Siamese Network with a margin value of 0.5. This algorithm surpassed the deep CNN based strong baseline algorithms.
Geometrical features including length of the chromosomes, end points etc. are used in the majority of research work towards chromosome feature extraction.69
Precisely, estimating the length of the chromosome; i.e. number of pixels along the centerline and, the location of the telomere, i.e. the edge of the chromatid are the two most common geometric features used in centromere detection.35,36 In addition, centerline detection is required to estimate the density profile.69
One common method of centerline detection found in early literature is skeletonization.77 However, this method fails because it produces spurious branches at the telomere due to the morphological variations present in the chromosomes. Therefore, applicability of this method is limited.35 Arachchige et al.36 addressed this issue by introducing a hybrid algorithm using GVF active contours, Discrete Curve Evolution based skeleton pruning and, morphological thinning. This algorithm was tested using 120 DAPI-stained lymphocyte cell images. The proposed algorithm was compared with gold standard using Mean Absolute Distance and Maximum Absolute Distance. The algorithm was able to detect centromere in bent chromosomes accurately. This approach can be improved by testing it with various cell types and increasing the number of samples in the dataset.
Generally, polarization is performed after the classification. Poletti et al.35 introduced a method to orient the chromosomes before the classification regardless of the reading orientation. This method reduced feature variance among the chromosomes in the same class and resulted in accurate classification. This research team worked with Q-band images. Using a large dataset of 5,474 chromosomes was a merit of this research. This dataset included amniotic fluid and choroidal villi cells. Robust estimation of the medial axis of the chromosomes was performed in order to extract the features, namely, length of the chromosome, density profile and contour function. It was assumed that the chromosomes were already segmented by using their previous work presented in 2009.38 The results were compared with the manual classification performed by an expert. This system showed 94% accuracy.35 However, manual classification by only one expert was subjective. Therefore, collecting data of manual classification from a group of experts could have justify the results in a better way.
A study was conducted by Vaurijoux et al. to detect di-centric centromeres.78 The results showed 50% misclassification due to the presence of multiple centromeres in the object despite the fast execution of the algorithm. However, it showed 4.35% accuracy than the manual process.49,78
The US patent, US8605981B2, addressed the drawbacks of manual and semi-automated centromere detection techniques, such as false positive detection and errors occur due to non-rigid chromosome boundaries. This algorithm had the capability to detect at least one centromere in each metaphase di-centric chromosome. The centerline was accurately drawn after proper segmentation of metaphase chromosomes. The number of centromeres was counted by identifying the longitudinal cross sections with minimum width by calculating the number of pixels with the same intensity. Accordingly, the percentage of di-centric chromosomes in a population of cells was counted. This was used to calculate the radiation dose using a previously determined dose-response curve from a calibrated source.60
The same research group developed an algorithm with higher accuracy to detect di-centric metaphase chromosomes to determine the radiation exposure. The dataset included overlapped and overspread chromosomes in a cell population. First, the overspread chromosomes were excluded by separating the segmented image into two depending on the area of the detected metaphase chromosome regions. The method of separating single metaphase chromosomes from chromosome clusters introduced by Rizvandi et al., was used to identify chromosome clusters. In order to do that, the algorithm approximated the centerline and counted the co-joints within the blob. The number of co-joints classified the blob into single chromosome or chromosome cluster. Clusters with multiple chromosomes were prone to be detected as false di-centric metaphase chromosomes. Therefore, the same procedure followed by Rizvandi et al.79 was used to separate touching and overlapping chromosomes. Next, the chromosomes were extracted using GVF which was superior to traditional active contour models because it used non-conservative forces in contouring. GVF could detect the concavities, hence the bent chromosomes. The centerline of each long chromosome and short chromosome was detected by incorporating cubic spline interpolation with Discrete Curve Evolution and Medial Axis Thinning36 respectively. Finally, centromeres were detected using the concept of minimal width. Another feature of this algorithm is parallelization, which improved the time efficiency. The proposed algorithm was applied only to chromosomes that were longer than a specific length where there was a high tendency to bend. To process the shorter chromosomes, a different thinning algorithm was used.80
In 2016, Subasinghe et al presented an advanced method that can handle large morphological variations to address the issue of artifacts occurring due to premature sister chromatid separation. Support Vector Machine was used to detect the telomere region. It accurately detected chromosomes with sister chromatid separation. This research work was presented mainly in three steps. First, the chromosomes were segmented and centerlines were extracted. Next, the contour was partitioned and chromosomes with sister chromatid separation were detected. The final step was centromere detection. This method showed 87% accuracy for a data set of 1400 chromosomes. It was a significant improvement compared to their previous attempt which achieved 81% accuracy with 226 chromosomes. The candidate Based Centromere Confidence (CBSS) was used to measure the confidence in each centromere detection. This metric might be helpful in improving time efficiency. In addition, this algorithm worked for both DAPI and Giemsa.7
Jahani and Setarehdan73 implemented an algorithm to straighten the chromosomes with an ability to detect single chromosomes in the entire image. Median filter was utilized to reduce the noise in the chromosome images. Next, the medial axis of the binarized image was extracted based on thinning. The medial axis was extended from both sides, assuming that the next 5 neighboring pixels had the same slope. A perpendicular line to the medial axis at each pixel was drawn. Finally, a vertical chromosome was obtained by making all the perpendicular lines lie parallel to each other. End-points of the medial axis were detected using morphological thinning and two masks. Then, a gray-level mask which varies the intensity level from 0 to 255 gradually, was used between the two end-points. According to the definition, the centromere is the narrowest area in the chromosomes. Accordingly, minimum total number of pixels having the same intensity was selected as the centromere.
Li et al.49 detected the centromeres of each isolated chromosome. Consequently, this research group was able to detect dicentric chromosomes. The maximum width of the chromosome was calculated by measuring point-wise inner distances. The areas that exceeded the pre-defined maximum width were removed as outliers. Centromeres were localized using a similar algorithm presented by Subasinghe et al.7 They implemented this algorithm using GVF, DCE, IIL, and SVM. Subsequently, di-centric centromeres were separated from monocentric chromosomes using a compound Machine Learning model. This model provided a solution for misclassifications occur when multiple centromeres present in one object. However, the accuracy achieved for Giemsa-stained images was comparatively low due to the presence of chromosomes with severe sister chromatid separation. In addition, staining debris resulted in over-segmentation. To address the aforementioned issues, conditional filters were introduced to the algorithm.49
The introduced algorithms for chromosome extraction by Mohommed,81 Jahani and Setarehdan,73 and Li in 2016,49 require smooth boundaries of chromosomes. However, chromosome boundaries are irregular, so the accuracy of centerline and centromere detection using the aforementioned algorithms should be verified by using irregular boundaries as well.
Madian et al.63 proposed a method to detect centromere in extremely bent chromosomes. First, the contours of the chromosomes were identified using GVF in a binarized image. To find the threshold value, the intra-class variance was minimized while increasing the inter-class variance. Curvature function and weighted shortest path calculation were used to identify the centromere. Medial Axis Transform (MAT) provided better results for straight chromosomes. For both straight chromosomes and bent chromosomes, the projection vector performed with more than 90% accuracy in detecting the centromere. The proposed algorithm provided accurate results for highly bent chromosomes comparatively.
To summarize the section, centromere detection is essential for the diagnosis of genetic disorders. Telomere detection and centerline detection lead to accurate centromere detection. However, morphological variations and weak boundaries are challenges during the process. The use of improved algorithms incorporating GVF, has proven promising solutions in feature extraction.
Table 3 presents a summary on selected state-of-the-art approaches82 towards chromosome segmentation and feature extraction.
| Publication | Method |
|---|---|
| Wang et al., 202183 | Adaptive Receptive field Multi-Scale network (ARMS Net) |
| Mei et al., 202068 | U-net & Conditional GAN |
| Bai et al., 202084 | U-net & YOLOv3 |
| Al-Kharraz et al., 20203 | YOLOv2 & VGG19 |
| Sale et al., 20194 | U-net |
| Qin et al., 201985 | G-Net & L-Net |
| Somasundaram et al., 20195 | CNN |
| Xie et al., 201986 | ResNet-50 |
| Swati et al., 201887 | Super-Xception network |
| Sharma & Vig, 201875 | ResNet-50 |
| Zhang et al., 201888 | CNN |
| Jindal et al., 201776 | Twin Siamese Network (CNN) |
Most of the chromosome segmentation and feature extraction algorithms are restricted to a particular type of staining. However, to fulfil the clinical requirement, algorithms which work despite the method of staining is required.
Working with high resolution images in digital image processing always provides better results. However, the methods that provide better visualization of chromosomes may result in significant chromosomal damage, consequently inaccurate diagnosis. Therefore, visualization methods that provide images with high resolution with negligible chromosomal damage are required.16,26,27 Nevertheless, algorithms might be improved using novel image processing concepts to work with low resolution and distorted chromosome images.89
One common limitation of the approaches towards chromosome segmentation and feature extraction is the undesirable smaller dataset. Hence, the reliability of these approaches remains low. Therefore, work with large dataset by using image augmentation, adding noise to the image and changing the scale is recommended to improve the diversity of the dataset.3
State-of-the-art human chromosome segmentation and feature extraction algorithms incorporate learning architectures. However, time efficiency of these algorithms depends on the complexity of the chromosome image. Precisely, presence of large clusters with overlapping and touching chromosomes reduces the overall time efficiency.28 New approaches should focus on architectures with’parallel run’ to improve the time efficiency as current development of the computer processors supports complex algorithms. In addition, selecting the optimum number of parameters for the learning architecture can minimize the execution time. There are existing reliable models such as AlexNet and ResNet which can be adapted and improved.89
The majority of the algorithms fail to disentangle both touching and overlapping chromosomes. In addition, some algorithms are incapable in working with bent chromosomes and di-centric chromosomes. However, algorithms which satisfy the clinical requirements should work with all of these types of chromosomes. Therefore, common approaches towards detecting touching, overlapping, bent and di-centric chromosomes are required. In addition, karyotyping is recommended for the suspicious cases of genetic disorders. Hence, translocations may present in the image which affect the banding pattern. Intelligent algorithms can be developed to address such cases. In addition, active learning may minimize the crowd dependency.6
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)