Keywords
MAF, Mutation, Single Nucleotide Variants, Visualization, Dashboard, WES, WGS
This article is included in the RPackage gateway.
This article is included in the Bioinformatics gateway.
MAF, Mutation, Single Nucleotide Variants, Visualization, Dashboard, WES, WGS
In the last decade, the cost of next-generation sequencing (NGS) has gone down exponentially as both throughput and novel methods continue to advance.1 For human clinical research, this has been reflected in an ever-growing number of datasets describing genomic variation among both normal and disease cohorts, including the 1000 Genomes Project Consortium,2 and the more recent gnomAD project,3 which still serve as important benchmarks of normal genomic variation in humans. Similar efforts for characterizing somatic mutations in cancer research have been completed for 33 tumor types by The Cancer Genome Atlas (TCGA) consortium,4 and over 1,700 cancer cell lines in the Cancer Cell Line Encyclopedia (CCLE) project from the Broad Institute.5 Although both TCGA and CCLE provide mult-omics data, single nucleotide polymorphisms (SNPs) and small insertion/deletions (Indels) from NGS data are often used as starting points for downstream analyses diving deep into the biological pathways and identifying drug target genes in these cancers.6
Both TCGA and CCLE provide somatic mutations freely as Mutation Annotation Format (MAF) files. This format is used to report high quality somatic variants for cohorts of cancer patients as it is more readable and portable than the traditional variant call format (VCF), and is therefore a common starting point for downstream analysis of somatic SNP/Indel data. R packages like ‘maftools’7 are frequently used by bioinformaticians to read, summarize, and perform statistical tests on data from MAF files, and they provide excellent functions for basic visualization, and flexible manipulation of the underlying data.
Since MAF files can contain a large number of annotations (e.g. the vcf2maf tool from MSKCC8 produces 136 columns of annotations with a default installation of Variant Effect Predictor9), selecting useful information and preparing it for discussion with researchers requires expertise. To simplify this task, we have developed MAFDash, an R package that helps to quickly create HTML dashboards for summarizing and visualizing data from MAF files. The resulting HTML file serves as a self-contained report that can be used to explore and share the results. MAFDash provides preset functions for extracting and organizing somatic variant data into interactive tables and figures. The goal of this package is to provide a simplified interface to filter and present data from MAF files suitable both for highly customized reports, as well as routine output from variant calling pipelines. The package also provides functions to generate individual plots as a ggplot210 or ComplexHeatmap11 object giving users more flexibility.
MAFDash is a package intended for use with the R programming language.12 The report is generated with a parameterized R Markdown script to arrange all the information. If a MAF object is provided, an interactive table is generated to provide client-side, dynamic filtering of the variant data. In addition to the dashboard generation, it also consists of a variety of functions to generate high quality figures to visualize mutation data. We also provided detailed documentation and a test dataset to demonstrate usage of these functions. Static plots are generated using the R packages ‘maftools’,7 ComplexHeatmap,11 ‘circlize’,13 and ‘ggplot2’.10 Interactive visualizations are implemented using ‘canvasXpress’14 and ‘plotly’.15
MAFDash was developed and tested on 2019 Macbook Pros with 2.4GHz 8-core Intel Core i9 processors and 16 Gb of memory, running Mac OS X 10.15.7 (Catalina). The source code and documentation is hosted on Github (https://github.com/CCBR/MAFDash).
The function getMAFdataTCGA(…) retrieves TCGA mutation data in MAF format. This function takes the cancer code(s) as input and outputs the TCGA mutation data called from Mutect2,16 or other callers as available. This function internally uses the ‘TCGAbiolinks’ R package17 to download the data and then uses internal processing to output the mutation data in a clean format. For annotation information, the getTCGAClinicalAnnotation(…) function extracts and processes common clinical features provided with the TCGA data including pathological state, tissue site, age, gender, race, and vital status, and generates reasonable preset colors suitable for use with ‘ComplexHeatmap’. The processed mutation data along with the clinical annotations can be further analyzed by utilizing the various visualization functions in MAFDash.
The filterMAF(…) function in MAFDash automatically detects the presence of relevant columns and re-casts them appropriately for numeric or text-based filtering. These include tumor read frequency and depth, frequency in population databases (gnomAD18 and ExAC19), and consensus mutation calls from multiple variant callers. Such criteria are frequently used for determining tumor mutational burden (TMB) from whole-exome sequencing data.20
This function also can also remove a preset list of commonly mutated genes,21 or a custom set of genes. Finally, data is processed in definable chunks of lines (default of 10,000 lines), which is intended to help filter large MAF files without getting “out of memory” issues.
MAFDash consists of various functions for visualizing summarized mutation data across a cohort of samples. Below are the different functions that are provided.
• generateBurdenPlot(…): It generates a dotplot and a barplot to show the comparison of the total number of mutations across the samples. The mutations are also grouped based on its type.
• generateMutationTypePlot(…): It generates a barplot showing the distribution of the silent and non-silent mutations across the input samples.
• generateOncoPlot(…): It generates a heatmap that summarized the top mutated genes across the input samples.
• generateOverlapPlot(…): It generates a circular plot to show the common mutations across the input samples.
• generateRibbonPlot(…): It generates a heatmap to show the cosine similarity between the mutated genes using the result from maftools’ somaticInteractions(…) function.
• generateTiTvPlot(…): It plots the frequency of transitions and transversions of the gene mutations in the input datasets.
• generateTCGAComparePlot(…): It computes and plots the mutation load of the input MAF against all 33 of the TCGA cohorts derived from MC3 project. It also calculates the significant mutational load differences between the cancers.
Mutational signature matrix for single-base substitutions (SBS) were retrieved from COSMIC v3.2.22 Text in the “Acceptance criteria” section of each signature page was retrieved from the COSMIC website using R scripts. This free text was lightly filtered and manually curated yielding 25 broad categories for 78 total signatures and is provided with the package repository in tabular format (Table 1).
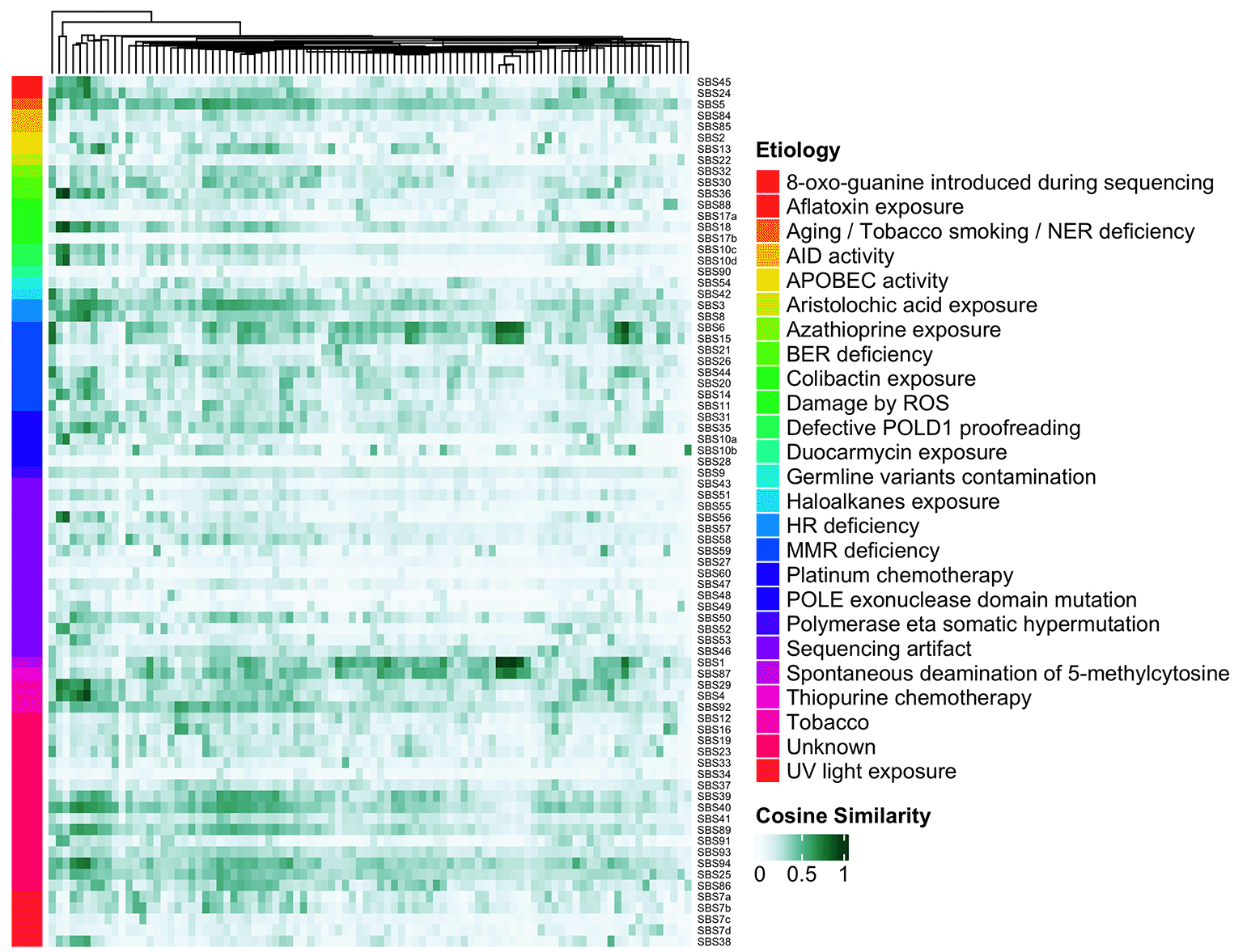
To aid interpretation of mutational signature analysis, we have curated COSMIC signatures etiologies from COSMIC v3.2.22 Specifically, we scraped the COSMIC website to retrieve the proposed etiology for all 78 COSMIC single-base substitution (SBS) signatures, yielding 36 unique etiologies, which we further manually curated into 25 broad categories. The generateCOSMICMutSigSimHeatmap(…) function shows these categorized proposed etiologies as colored row annotations, aimed at quickly identifying distinct or common etiologies across a cohort. Figure 1 shows the SBS signature in each sample in columns, COSMIC mutation signatures in rows, and each cell is colored to indicate the level of similarity between the two.
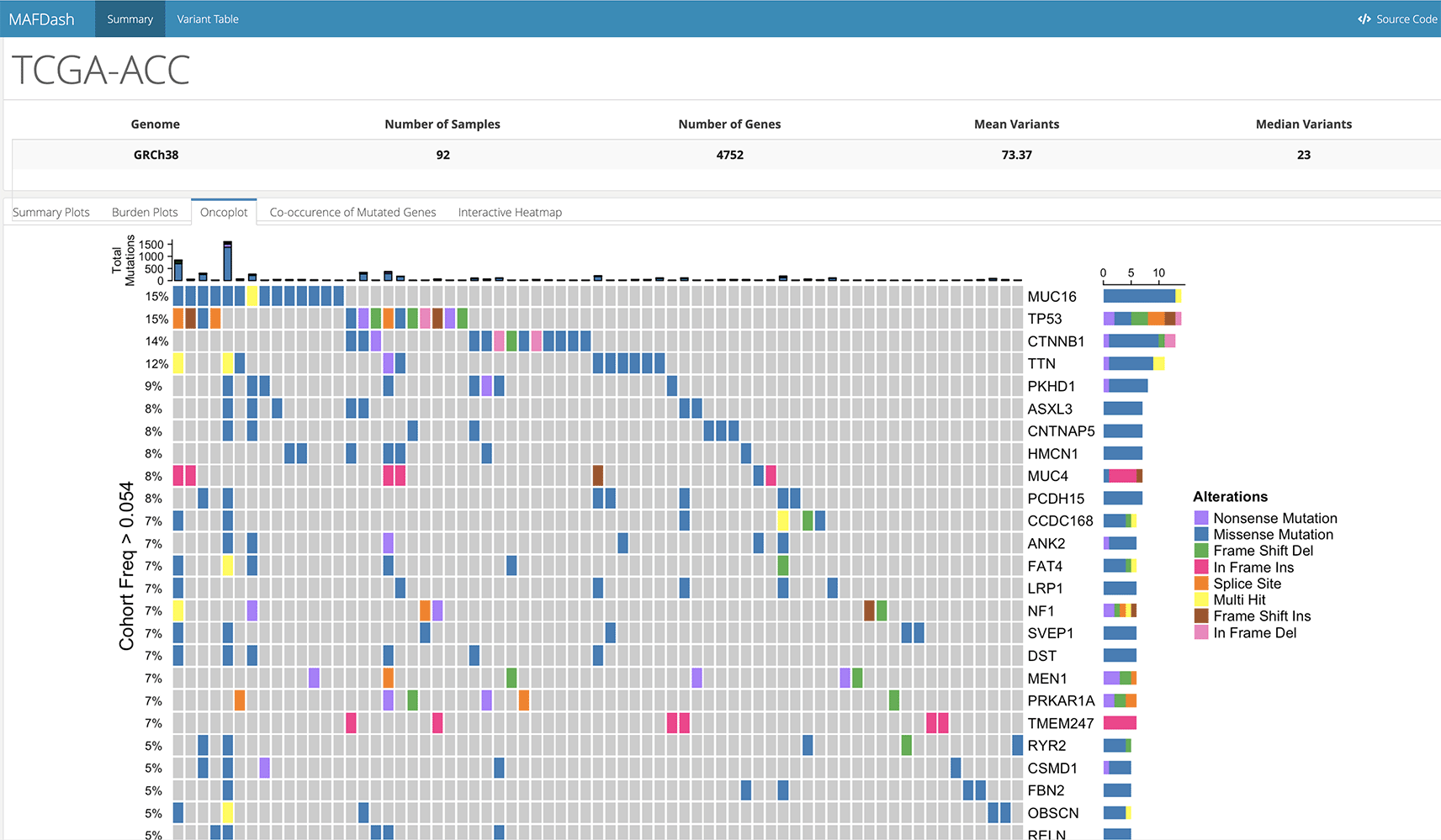
MAFDash has a function (getMAFDashboard(…)) that generates an HTML dashboard for visualization and analysis of mutation data in MAF format. The dashboard consists of arbitrarily defined or preset interactive plots describing the data. By default, if MAF data is provided, the dashboard visualizes the mutations data in five different tabs.
• Summary plots: Static multi-part figure describing cohort summaries of variant classification, variant type, number of variants per samples and nucleotide change (from ‘maftools’).
• Burden plots: Interactive plots showing the number of variants per samples in the form of a dotplot and barplot, with hover text containing sample and mutation information.
• Oncoplot: Plot summarizing the top mutated genes across the samples.
• Co-occurrence of mutated genes: A circular ribbon plot showing co-occurrence of the mutations, inspired by the somaticInteractions(…) function in ‘maftools’.
• Interactive heatmap: An interactive version of the oncoplot with hover text showing the number of mutations in a gene for a particular sample.
In addition to these plots, an interactive table is generated using the DT23 and crosstalk24 R packages to provide client-side, dynamic filtering of the variant data. The generated dashboard is self-contained for sharing with collaborators. MAFDash will automatically account for missing data and also provides reasonable defaults for filtering mutation data. Figure 2 shows the dashboard output for Adrenocortical carcinoma (ACC) downloaded from TCGA.6
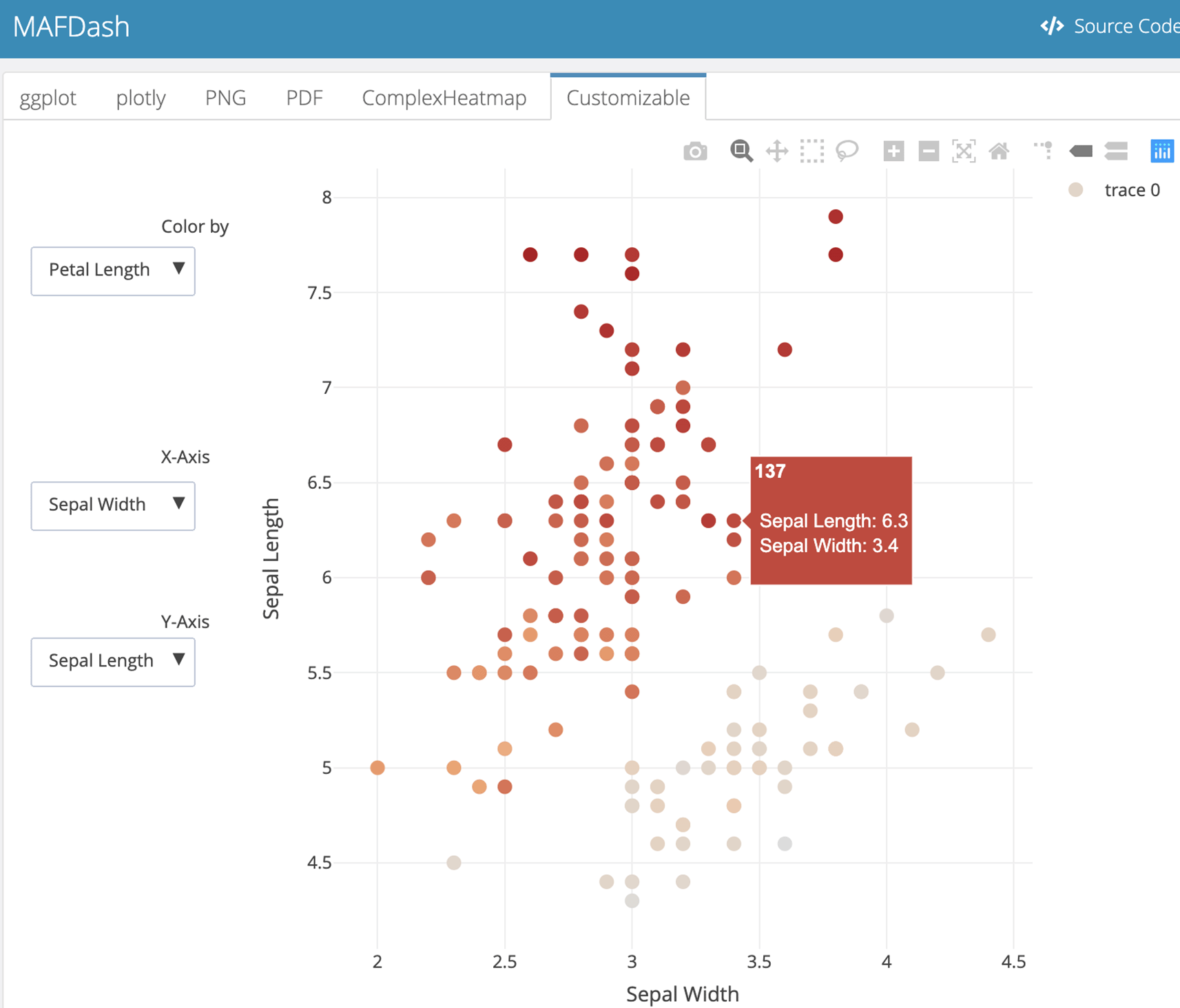
Even without MAF data, MAFDash can be used to generate an HTML report with user generated plot objects. Users can pass any ‘ggplot2’, ‘ComplexHeatmap’, or ‘plotly’ objects, or the location of an image file to include it in the dashboard as a list, and have it rendered as a dashboard with each element as a tab in the report. Figure 3 shows an example dashboard using the iris dataset provided with R.
We developed MAFDash to simplify the process of generating interactive reports for somatic mutation analysis. The ‘maftools’ R package already provides a comprehensive toolkit for organizing and analyzing MAF data, but it exclusively uses base R graphics for plotting, which is not amenable to further modification or interactivity. For example, the tcgaCompare(…) function is an excellent visual comparison of mutation burden with all cancer types in TCGA. To allow interactivity in MAFDash, we implemented the same visualization using ‘ggplot2’, which can trivially be converted to an interactive HTML widget using ‘plotly’. Finally, the self-contained nature of the HTML report, as well as a range of choices for interactive plots, is aimed at easily sharing data and interpretations. Overall, we hope that MAFDash will allow for quick iterations of analysis during collaborations between bioinformaticians and bench scientists.
All data underlying the results are available as part of the article and no additional source data are required.
• Source code available at: https://github.com/CCBR/MAFDash
• Archived source code at time of publication: https://doi.org/10.5281/zenodo.642183325
• License: MIT License
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)