Keywords
Pharmacometrics, pharmacokinetics, pharmacodynam- ics, software, R, package
This article is included in the Bioinformatics gateway.
This article is included in the RPackage gateway.
Pharmacometrics, pharmacokinetics, pharmacodynam- ics, software, R, package
Pharmacometrics (PMx) is the discipline of the description and quantification of interactions between xenobiotics and patient’s biologic system (human and non-human) using mathematical models and taking into account both beneficial and adverse effects1. These models are used in various circumstances, such as supporting R&D drug development in the pharmaceutical industry2, especially through the implementation of Model-Informed Drug Discovery and Development (MID3)3, or in hospitals for therapeutic drug monitoring4. In addition to the creation of the analysis plan and the Pharmacometrics report, the typical operational workflow of Pharmacometrics analyses includes: 1) pre-modeling work with data assembly, followed by graphical and statistical exploration, 2) iterative core modeling work with equation writing, parameter estimation, and model evaluation, and 3) final model exploitation, mostly through simulations, computation of secondary parameters, and/or parameter interpretation for the patients, in order to inform decision-making for a new study or individual patient dosing5,6.
Pharmacometricians routinely use a variety of tools ranging from generic programming software (e.g., R) to specific PMx software (e.g., NONMEM7, Monolix8, Phoenix WinNonLin9, in addition to multiple dedicated R packages). These PMx programs’ performance can be assessed both in terms of efficacy and efficiency. Efficacy is the ability of software to produce a desired or intended output, while efficiency represents the resources (time, effort) required to generate the intended output10. The efficacy of most of the PMx software is satisfying, thanks to the variety of available programs and the flexibility that they allow. Efficiency, on the other hand, can be improved, through a simplification of the model-coding or the improvement of interoperability, since switching from one software to another with the same model is standard practice to complete a PMx project, but most of the software uses different syntax for coding the same elements (e.g., structural equation or model results). This interoperability issue has already been reported, both for the overall workflow11 or specific tasks such as optimal design12.
Several projects have been created in various fields to improve or create interoperability between software, with at least two different paradigms showing successful results. In the first one, a new robust and generally marked-up language is created, aiming to become the standard of use. It has been created for system biology with the SBML project13. The Drug Disease Model Resources (DDMoRe) project (https://www.ddmore.foundation) has implemented this methodology in pharmacometrics14–17.
The second methodology consists of creating wrappers and translation tools from existing languages. The most successful and used tool of that kind is probably pandoc, dealing with document writing18. It allows notably to automatically translate and produce a LaTeX-based PDF, HTML or Word document (high efficacy but low efficiency) by simply coding in markdown (low efficacy but high efficiency syntax). The addition of a light translating tool between the two turned the system both highly effective and efficient. Looking at the markdown / (LaTeX) / PDF workflow, at least four main advantages of this system can be described:
1. It is not needed to know LaTeX to produce a LaTeX-based PDF document (reduced required knowledge).
2. It is easier and faster to write first a markdown file and translate it rather than directly coding in LaTeX (efficiency improvement at a single software level).
3. There is no additional work needed to produce an HTML or Word document if those formats are finally required (multiple software / interoperability efficiency improvement).
4. If needed, it is possible to extract the intermediary LaTeX file to verify and eventually modify the output (as a result, even imperfect or incomplete pandoc task often remains useful).
We developed Peccary, a new set of tools (R console package with its Shiny App19) that aims to provide the four previously mentioned pandoc benefits but in the pharmacometrics field. The goal is thus to gather and create links between pre-existing PMx software through various translating systems. R was judged the most appropriate platform for such a project because 1) it is the most used generic programming software in PMx11, 2) almost every R packages are under free licenses, meaning their source code are available and users can freely modify or redistribute them20,21, and 3) R packages already cover most of PMx tasks, such as a non-compartmental analysis (e.g., pknca22), model diagnostic (e.g., Xpose23, npde24 or vpc25), optimal design (e.g., PFIM26 and PopED27), simulation (e.g., simulX28), parameter estimation (e.g., nlmixr29 or saemix30) and linker to proprietary software (e.g., popKinR to manage NONMEM runs31 or lixoft-connector to control Monolix32). Having all these packages reunited in the same platform, such as Peccary, would therefore highly increases PMx efficiency.
Development process
Software can be developed through various methods. In 1999, Eric Raymond published an influential essay in which he compared software built as a cathedral to those built as a bazaar33. Briefly, software built as a cathedral are very well thought out and structured before any code integration whereas bazaar systems imply much less preparation but need an organic development through immediate code creation and aggregation.
Peccary was developed with the bazaar methodology principles. Concretely, Peccary’s development followed the main author’s daily work as a pharmacometrician. Whenever a specific need to produce an output was encountered, an immediate solution was coded in R, most of the time (but not always) trying to build a generic function with a metaprogramming system. Peccary is the aggregation of these snippets of R-code. This agile methodology allowed us to develop a consistent R package piece by piece, ensuring that each implemented feature is truly needed and tested in real life. Since Peccary’s goal is to improve the efficiency of PMx, all attention was given to optimize the time and number of steps required to produce each output.
Software availability
The source code availability of Peccary and its subpackages is described in the software availability statement.
The downloading and installation of Peccary (and all subpackages) is made directly in R using the following code:
devtools::install_github("Peccary-PMX/Peccary")
Peccary and its Shiny App have been tested both on Windows, mac OS and Linux (Arch distribution).
Overview of the workflow
The current structure of Peccary, depicted in Figure 1, comprises three subpackages. Briefly, PeccAnalysis deals with pre-modeling work, PeccaReverse with model building and simulation, and PeccaResult with model diagnostics. Peccary is the main package that gathers these three subpackages under one platform usable both in R console or inside a Shiny App.
Inside the Shiny App, users can create a Peccary’s project, allowing to save in the same place metadata of both the datasets (their path, separator, decimal, and “NA” strings) and various analysis (e.g., exploratory plots or model simulations). This system allows the analyst to reload any desired datasets when reopening the Shiny App easily, switch from one dataset to another with a single action, or reload and secondarily modify any pre-saved data analysis. Of note, Peccary requires the use of tidy data34, secondarily modifiable in the Shiny App.
Metaprogramming system
Most Peccary functions have been metaprogrammed35, meaning a Peccary independent R code/expression is produced before generating the outputs. This system was conceived to make Peccary useful both during the exploratory work phases (trying many things) and during the cleaning / reporting phase (selecting a few key analyses to report). Indeed, most of the produced works in a project are generally never published because they are just intermediate results in a trial-and-error workflow. During this exploratory phase, the Shiny app can directly produce the desired outputs, similarly to a direct markdown / PDF shortcut, with fast but unverified results that the user can only informally trust. However, every time a report integration needs to be made, the corresponding R code can be copy-pasted in an R/Rmd file before being further controlled (quality control) and eventually modified. Of note, this dual-system can dynamically co-exist, for Peccary is especially efficient working in a dual-screen set-up with two parallel R sessions - one using the Peccary Shiny app, the other a free R session for direct code transfer and manipulation.
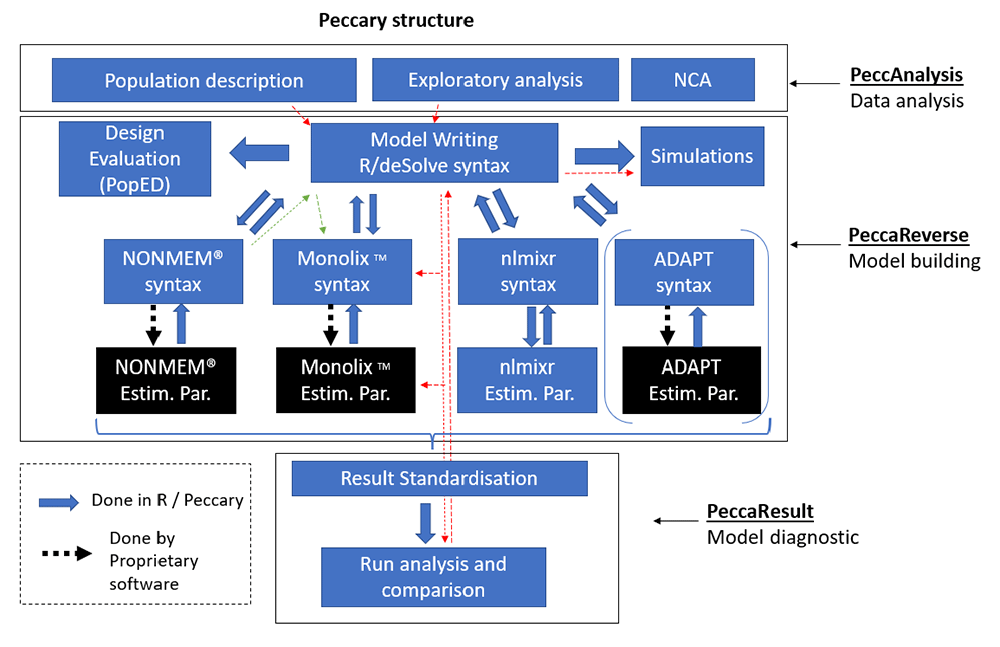
PeccAnalysis allows performing pre-modeling work such as population description, exploratory analysis and non-compartmental analysis (NCA). PeccaReverse allows to create or import a model with a minimal syntax, before using it for simulations, design evaluation, or translation into other syntaxes. Finally, PeccaResult standardizes and compares runs from multiple software with various analysis functions. Peccary can be used during a whole PMx process (red arrows) or just as a toolbox for any specific task (e.g., model translation from NONMEM to Monolix in green arrows).
PeccAnalysis
PeccAnalysis gathers all functions made to analyze the data before any modeling work. This mostly includes three different activities, namely 1) population description, 2) plot exploration, and 3) non-compartmental analysis (NCA).
The theophyline dataset pre-existing in R is used for demonstrative purpose, with the addition of the following columns: dose level (DL) as the rounded real-dose, the presence or absence of a side effect (true or false), and the gender (men or women) leading to Table 1:
demoDF <- as_tibble(Theoph) %>% mutate(DL = round(Dose), Subject = as.double(as.character(Subject))) %>% left_join(tibble(Subject = 1:12, side_effect = sample(c(T, F), 12, replace = T), gender = sample(c("M", "W"), 12, replace = T))) kable(head(demoDF))
Wt stands for weight, conc for drug concentration and DL for dose level. Gender values are M for men and W for women.
| Subject | Wt | Dose | Time | conc | DL | side_effect | gender |
|---|---|---|---|---|---|---|---|
| 1 | 79.6 | 4.02 | 0.00 | 0.74 | 4 | TRUE | M |
| 1 | 79.6 | 4.02 | 0.25 | 2.84 | 4 | TRUE | M |
| 1 | 79.6 | 4.02 | 0.57 | 6.57 | 4 | TRUE | M |
| 1 | 79.6 | 4.02 | 1.12 | 10.50 | 4 | TRUE | M |
| 1 | 79.6 | 4.02 | 2.02 | 9.66 | 4 | TRUE | M |
| 1 | 79.6 | 4.02 | 3.82 | 8.58 | 4 | TRUE | M |
Population dataset description
The population description generally requires two steps. The first step is to extract covariate information from the full dataset. Because PK/PD datasets generally contain multiple rows per ID (one for each observation and one for each drug administration), extracting only the covariate information as a reduced dataset is needed. This has been simplified with a function (pecc_search_cov()), automatically creating a covariate table by only inputting the column names representing the individuals (column “Subject” in our example). This function extracts only the columns with a unique value for each individual, thus considered covariates. After this dataset covariate extraction, descriptive statistics need to be computed and organized into a descriptive table, both operations already performed by the table1 R package36.
Overall, this whole process of population description has been embedded into a single PeccAnalysis function (pecc_table1_original()). In the Shiny App, where it is not needed to know the function’s name, the metaprogrammed R code is provided in addition to its corresponding table. The user can choose the output format in the console: either the R code, the final table, or both (through outputExpr argument). An example of an automatic covariate extraction and statistical description by providing only the source data frame, the name of the column representing the individuals, and an optional covariate name to create the columns (other than “Overall”) is provided leading to Table 2:
pecc_table1_original(df = demoDF, col1 = DL, reduceBy = Subject, outputExpr = "Both") ## table1::table1(~Wt + Dose + side_effect + gender | DL, data = demoDF %>% ## distinct(Subject, Wt, Dose, DL, side_effect, gender))
Wt stands for weight, M for men and W for women. The corresponding metaprogrammed code was printed before its evaluation.
Of note, the “. . . ” argument (non-used above) allows the user to manually choose the desired covariates to display (instead of printing them all) but also to transform the numeric covariates into factors, even though the main interest of this function relies on its ability to detect the covariates automatically.
Specific case of Boolean covariate
If a covariate is a Boolean (only possible values are TRUE/FALSE or 0/1), a specific function counts how many patients have this covariate equal to TRUE (or 1) in regards to one or two other covariates. For instance, this is used to analyze the number of good responder patients to treatment, or, as in the following example, for counting the number of patients with side effects in regard to their genders and dose levels leading to Table 3:
pecc_search_cov(demoDF, Subject) %>% # the function to extract cov pecc_count(metric = side_effect, col_cov = gender, row_cov = DL ) %>% kable()
Gender values are M for men and W for women.
| DL/gender (side_effect) | M | W | Total |
|---|---|---|---|
| 3 | 0/1 | - | 0/1 |
| 4 | 1/3 | 1/1 | 2/4 |
| 5 | 3/4 | 1/1 | 4/5 |
| 6 | 1/1 | 1/1 | 2/2 |
| Total (0 NA) | 5/9 | 3/3 | 8/12 |
Of note, this function is one of the few from PeccAnalysis that is not yet metaprogrammed. The first attempts led to a too cumbersome R code compared to the easiness of manually checking the results.
Graphical exploration of longitudinal data
Another PeccAnalysis function allows visualization of longitudinal data via appropriate ggplot objects. The function includes most of the classical ggplot customization (coloring observations through a covariate, performing subplot via wrap functions. . . ). Additional features have been added, such as a direct output standardization by a continuous column, or an improved wrapping system where other profiles are printed in grey background (improving the comparisons between groups). The following code displays both the metaprogrammed code and its results (Figure 2):
plot_spagh(demoDF, Time, conc, group = Subject, facetwrap = DL, col = gender,bkgrdalpha = 0.6, output = "Both", ylabs = "Concentration (units)") ## demoDF %>% filter(!is.na(conc)) %>% { ## explo_post_treatment <<- . ## } %>% ggplot() + geom_line(data = explo_post_treatment %>% mutate(peccaryTemp = ## paste0(group = Subject, ## col = gender)) %>% select(-DL), aes(x = Time, y = conc, group = peccaryTemp), ## col = "darkgrey", alpha = 0.6) + geom_point(aes(x = Time, ## y = conc, col = factor(gender)), alpha = 1) + geom_line(aes(x = Time, ## y = conc, group = Subject, col = factor(gender)), alpha = 1) + ## facet_wrap(~DL, labeller = label_both, scales = "free") + ## scale_y_log10(labels = labels_log, breaks = breaks_log) + ## labs(col = "gender", y = "Concentration (units)") + theme_bw(base_size = 15) + ## theme(plot.title = element_text(hjust = 0.5), plot.subtitle = element_text(hjust = 0.5), ## plot.caption = element_text(hjust = 0, face = "italic", ## colour = "grey18"))
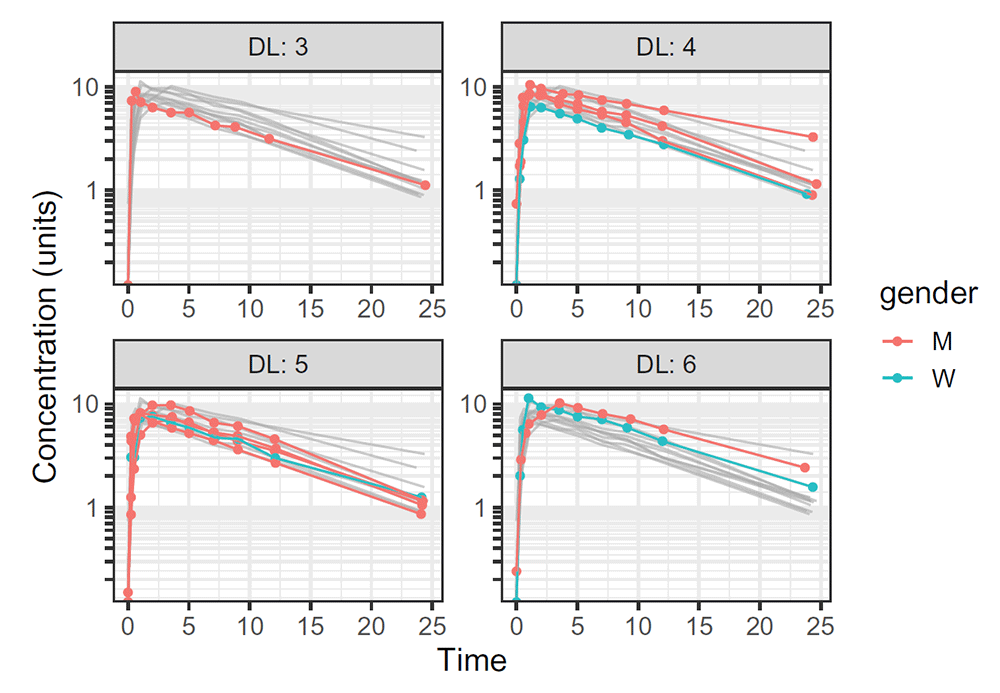
The corresponding R expression was printed before its evaluation.
An example of using this function inside the Shiny App is presented in Figure 3, where both the plot, the peccary dependent code (the plot_spagh(. . . ) code as shown above) and the peccary independent code (metaprogrammed R expression) are displayed simultaneously.
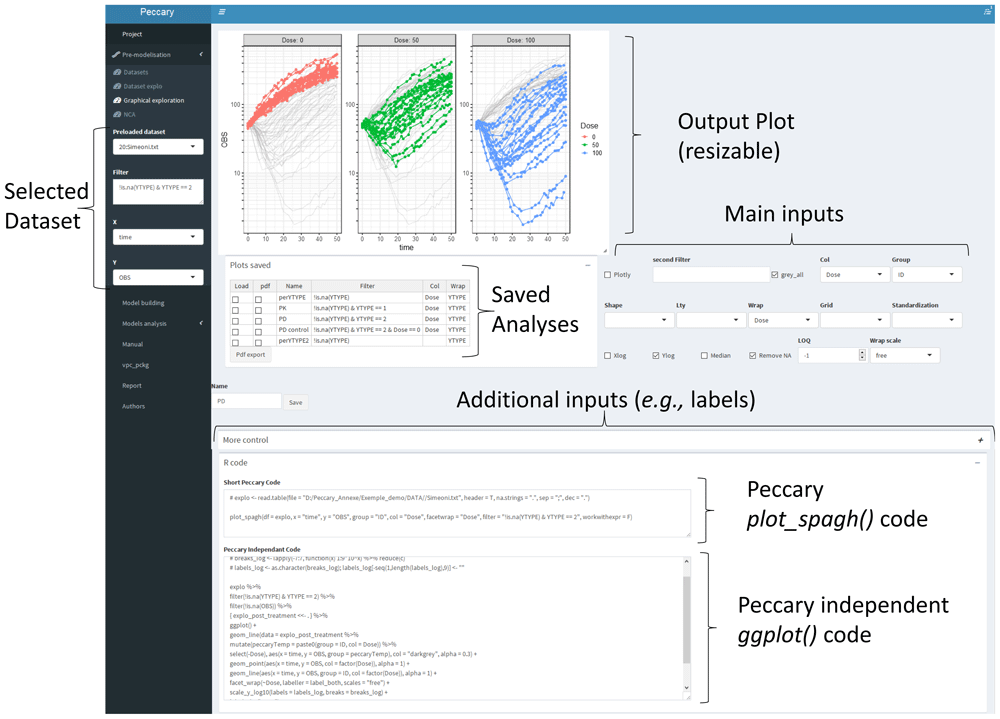
The inputs, produced plot, and metaprogrammed code (both Peccary dependent and Peccary independent) are displayed on the same page. In addition, each dataset can store the metadata of each analysis, such as a plot that can be recreated when reopening the Shiny app, before eventually being re-modified. On the left panel, the user can efficiently switch from one dataset to another (among the ones stored in a project or inputted since the beginning of the session).
Non-compartmental analysis (NCA)
Peccary simplifies the use of the pknca package22 for performing NCA in several ways. As a first step, the function automatically creates the dose dataset, either using an EVID column, or assuming dose administration at time 0 (depending on the inputs). Second, it simplifies the computation of AUC from time 0 to X (X being a numeric argument) by using the pknca “intervals” system. Integrating “intervals” also makes it easier to toggle the computation of more NCA parameters by modifying the metaprogrammed code (for instance setting vss.last to True in the following example). Third, it automatically transforms the final pknca results into an organized data frame, directly including all possible covariates (extracted with pecc_search_cov()) for further analysis. Results are shown in Table 4.
demoNCA <- peccary_pknca(demoDF, Time, conc,Subject = Subject, dose = Dose, AUC0_x = 24, computeMedian = F, outputExpr = "both") ## NCA_eval <- { ## conc_obj <- PKNCAconc(data = demoDF, formula = conc ~ Time | ## Subject) ## dose_obj <- PKNCAdose(data = demoDF %>% group_by(Subject) %>% ## slice(1) %>% mutate(`:=`(Time, 0), `:=`(conc, 0)), formula = Dose ~ ## Time | Subject) ## intervals_manual <- data.frame(start = 0, end = c(24, Inf), ## cmax = c(F, T), tmax = c(F, T), auclast = c(T, T), aucinf.obs = c(F, ## T), cmin = c(F, T), tlast = c(F, T), vss.last = c(F, ## F), cl.obs = c(F, F)) ## data_obj_automatic <- PKNCAdata(conc_obj, dose_obj, intervals = intervals_manual) ## pk.nca(data_obj_automatic) ## } ## left_join(NCA_eval$result %>% filter(end != 24) %>% select(Subject, ## PPTESTCD, PPORRES, end) %>% distinct() %>% spread(key = PPTESTCD, ## value = PPORRES), NCA_eval$result %>% filter(end == 24) %>% ## select(Subject, PPORRES) %>% rename(`:=`(AUC24, PPORRES))) %>% ## left_join(demoDF %>% distinct(Subject, Wt, Dose, DL, side_effect, ## gender)) demoNCA[1:5, c("Subject","gender","cmax", "auclast", "AUC24", "tmax")] %>% kable
| Subject | gender | cmax | auclast | AUC24 | tmax |
|---|---|---|---|---|---|
| 1 | M | 10.50 | 147.23475 | 92.36544 | 1.12 |
| 2 | M | 8.33 | 88.73128 | 67.23456 | 1.92 |
| 3 | M | 8.20 | 95.87820 | 70.58886 | 1.02 |
| 4 | M | 8.60 | 102.63362 | 72.84350 | 1.07 |
| 5 | W | 11.40 | 118.17935 | 84.39951 | 1.00 |
Of note, a very similar function (peccary_pknca()) metaprograms the NCA computation from scratch, manually computing all classical NCA parameters that do not require extrapolation (Cmax, Tmax, AUCTlast,. . . ).
# demoNCA <- peccary_NCA(demoDF, timecol = Time, obscol = conc, Subject, auc_0_X = 24)In both cases, a table with NCA individual parameters is computed. In the Shiny app, a table1 function summarizes the results by possible covariates, along with the generation of either a boxplot or a scatter plot depending on the specific covariate the user wants to analyze. Those plots are generated using the GGpubr package37, directly integrating statistical tests. The following code generates boxplots (Figure 4) using NCA results:
plot_boxplot(df =demoNCA, x = gender, auclast, cmax, tmax, AUC24, outputExpr = "both", methodCompar = "wilcox.test") ## demoNCA %>% group_by(gender) %>% mutate(`:=`(gender, paste0(gender, ## "\n(n=", length(gender), ")"))) %>% gather(auclast, cmax, ## tmax, AUC24, key = "key", value = "value") %>% ggplot(aes(factor(gender), ## value)) + geom_boxplot() + geom_point() + facet_wrap(~key, ## scales = "free") + ggpubr::stat_compare_means(comparisons = list(c("M\n(n=9)", ## "W\n(n=3)")), method = "wilcox.test") + scale_y_log10() + labs(x = "gender") + ## theme_bw()
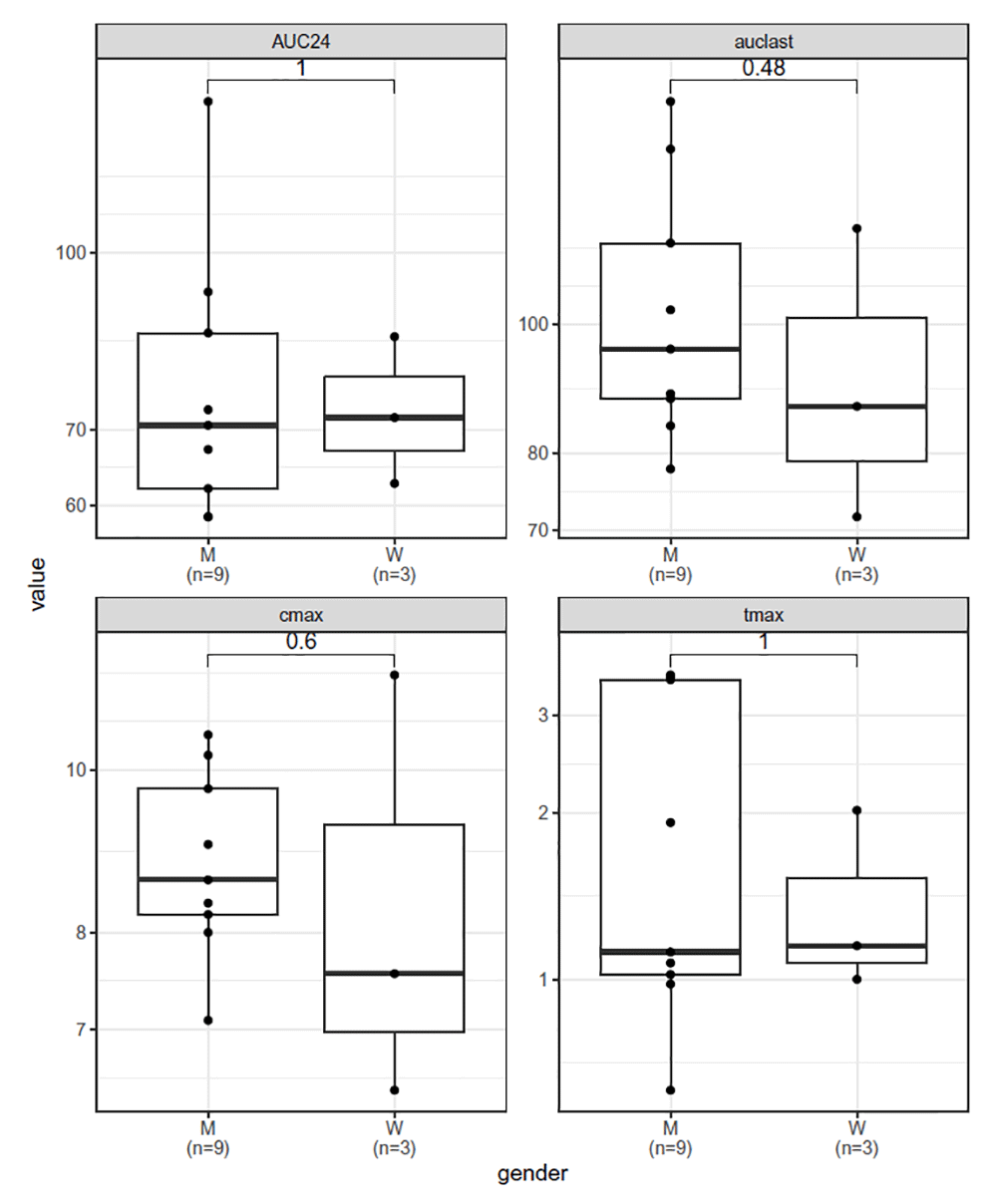
Of note, plot_boxplot() is a generating boxplot function that can be used in other circumstances. All auclast,cmax, tmax, and AUC24 belong to the “. . . ” arguments, allowing to add more parameters in the facet_wrap() code.
The following code produces a scatter plot (Figure 5) to visualize the relationship between a continuous covariate and NCA parameters:
plot_correlation(df = demoNCA, x = Wt, auclast, cmax, tmax, AUC24, outputExpr = "both", cor.method = "spearman" ) ## demoNCA %>% gather(auclast, cmax, tmax, AUC24, key = "key", value = "value") %>% ## ggscatter(x = "Wt", y = "value", add = "reg.line", cor.coef = TRUE, ## cor.method = "spearman", conf.int = FALSE) + theme_bw() + ## facet_wrap(~key, scales = "free") + scale_y_log10() + scale_x_log10() + ## labs(x = "Wt")
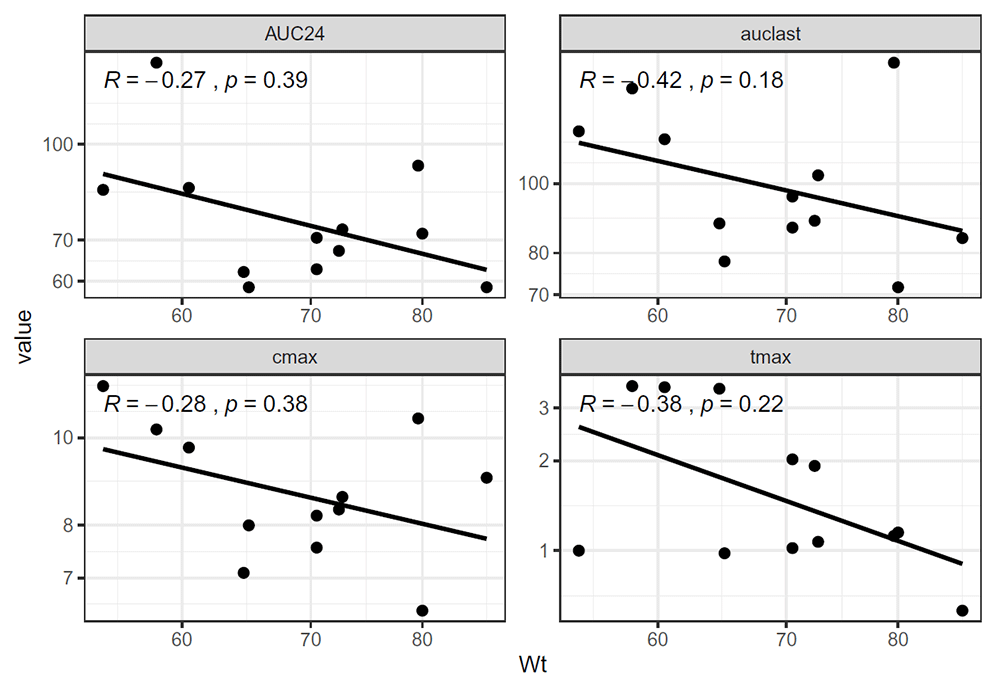
Of note, plot_correlation() is a generating scatter plot function that can be used in other circumstances. All auclast, cmax, tmax, and AUC24 belong to the “. . . ” arguments, allowing to add more parameters in the facet_wrap() code.
The Shiny app gathers all metaprogrammed R codes in a single bloc (NCA generation, table1 generation, and covariate analyses).
PeccaReverse
PeccaReverse is the second Peccary subpackage whose role is to simplify model creation, dynamic exploration, simulations, and translation into other software syntaxes. A simplified deSolve syntax has been used as the core language of PeccaReverse. For example, to create two compartments for a PK model with oral drug absorption, one can simply write the structural model as follows:
model_demo <- "dGut <- - ka * Gut dCentral <- ka * Gut + Periph * k21 - Central * k12 - ke * Central dPeriph <- Central * k12 - Periph * k21 conc_output <- Central/vd"
Peccary also contains a PK/PD library to avoid writing frequently used models from scratch. Then, the deSolve_pecc() function automatically extracts all the model compartments, parameters, and initial states (if provided) from the equations, and also reconstitutes both deSolve and RxODE expressions:
deSolve_pecc(model_demo) ## $model ## function(t, state, parameters) { ## with(as.list(c(state, parameters)), { ## dGut <- -ka * Gut ## dCentral <- ka * Gut + Periph * k21 - Central * k12 - ## ke * Central ## dPeriph <- Central * k12 - Periph * k21 ## conc <- Central/vd ## list(c(dGut, dCentral, dPeriph), c(conc = conc)) ## }) ## } ## ## $modelrxode ## RxODE({ ## d/dt(Gut) <- -ka * Gut ## d/dt(Central) <- ka * Gut + Periph * k21 - Central * k12 - ## ke * Central ## d/dt(Periph) <- Central * k12 - Periph * k21 ## conc <- Central/vd ## }) ## ## $state ## [1] "Gut" "Central" "Periph" ## ## $parameter ## [1] "ka" "k21" "k12" "ke" "vd" ## ## $output_manual ## [1] "conc" ## ## $toplot ## character(0) ## ## $initialCond ## # A tibble: 0 x 2 ## # ... with 2 variables: Cmt <chr>, value <chr>
Briefly, deSolve_pecc() function works by analyzing the text through regular expression manipulations (grep, grepl, gsub, . . . ). Everything not recognized as a compartment (here, Gut, Central and Periph, recognized with the initial “d” and some other conditions to distinct them from variables starting with “d”), a computed variable (every word before “<-”, here conc), or a reserved word (such as log, exp, if, else . . . ) is directly considered as a parameter (ka, k12, k21, ke and Vd). Adding “_output“ or “_plot“ (automatically removed in the produced deSolve/RxODE code) at the end of a variable means this variable is respectively an output of the model or that it needs to be reported after simulations for plotting.
Within the Shiny app, once the structural model has been analyzed (through an action button), various input tables are automatically created and pre-filled, as depicted in Figure 6. Filling the missing parts of these tables (further described) allows the users to perform most of PeccaReverse tasks, namely 1) simulations, 2) design evaluation, and 3) various syntax translations. The following sections describe the main console functions internally invoked while using the Shiny App. It is important to note that this system is much less efficient in console mode because it requires writing input tables from scratch, plus the need to know how to use the internal functions (compared to a more intuitive and dynamic use in the Shiny app).
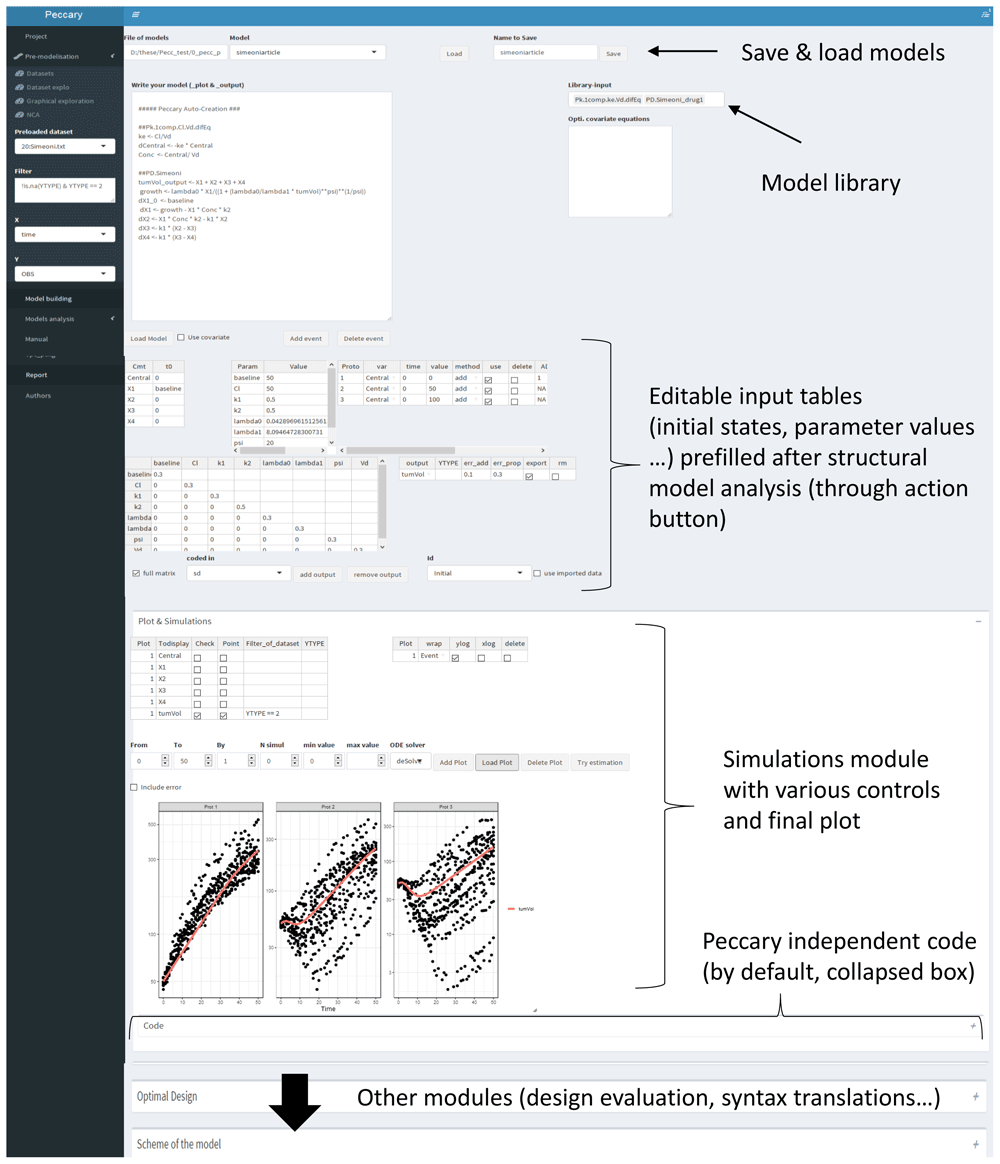
The model equations are first written (or imported from the model library) in the top left bloc. After clicking on an action button below the equations, the model is analyzed, and various input tables are created and pre-filled. After completing these tables (e.g., attributing a value for each parameter value), the user can perform simulations in the corresponding module, here displaying a tumor growth inhibition model with three different dose levels. On each panel, the corresponding observations were added. The whole Peccary independent code for performing this plot can be seen below the plot (by unrolling the collapsed box). All other PeccaReverse modules (design evaluation, syntax translation, . . . ) are in the other collapsed boxes below on the same page.
Simulations
Performing simulations and plots was divided into three steps: 1) programming the parameters sets, 2) programming the simulations, and 3) programming the plots. For each step, several methods exist, leading to multiple possible combinations. The metaprogrammed R code is incremented after each of the three steps to produce the final R code.
The first step consists of programming the sets of parameter values to simulate. It requires at least one table containing the population values, the distributions (normal, log-normal) and the information on whether each parameter is estimated or fixed. Importantly, several values can be attributed to a parameter to perform a local sensitivity analysis automatically. The user can choose between simulating those exact values or sampled values within a distribution. A variance/covariance matrix (coded in standard deviation or variance) should be inputted in the late case. Finally, a function generates the code for sampling parameters sets, in the following example asking for 100 parameters sets for three possible k21 values:
# Parameter value table param_df <- tribble(~Param, ~Value, ~ Distrib, ~E, "ka", 0.3, "logN", "Esti", "ke", 0.1, "logN", "Esti", "k12", 0.3 ,"logN", "Esti", "k21", c(0,0.1,0.3) ,"logN", "Esti", "vd", 5 ,"Norm", "Esti") # Variance/covariance matrix (here coded in sd) matrix_sd <- tribble(~ ka,~ k21,~ k12,~ ke,~ vd, 0.3,NA,NA,NA,NA, #ka 0,0.3,NA,NA,NA, #k21 0,0,0.3,NA,NA, #k12 0,0,0,0.3,NA, #ke 0,0,0,0,0.3 #vd ) # Final function for parameter sampling paramInd <- random_etas(n = 100,param_df , matrix_sd, sd = T, returnExpr = T) expr({!!!paramInd}) # (paramInd is a list of expr) ## { ## parameters <- tibble(ka = c(0.3, 0.3, 0.3), ke = c(0.1, 0.1, ## 0.1), k12 = c(0.3, 0.3, 0.3), k21 = c(0, 0.1, 0.3), vd = c(5, ## 5, 5), nameparset = c("k21 = 0", "k21 = 0.1", "k21 = 0.3" ## )) ## matrix_eta <- tibble(k12 = c(0.09, 0, 0, 0, 0), k21 = c(0, ## 0.09, 0, 0, 0), ka = c(0, 0, 0.09, 0, 0), ke = c(0, 0, 0, ## 0.09, 0), vd = c(0, 0, 0, 0, 0.09)) %>% as.matrix ## eta <- matrix(rnorm(100 * nrow(matrix_eta)), ncol = nrow(matrix_eta)) %*% ## chol(matrix_eta) ## eta <- apply(eta, 2, function(x) rep(x, nrow(parameters))) ## ind_param <- map_dfc(parameters, ~rep(.x, 100)) %>% arrange(nameparset) ## parameterslogN <- c("ka", "ke", "k12", "k21") ## ind_param[parameterslogN] <- ind_param[parameterslogN] * ## exp(eta[, parameterslogN]) ## parametersNorm <- "vd" ## ind_param[parametersNorm] <- ind_param[parametersNorm] + ## eta[, parametersNorm] ## }
Of note, no distribution sampling would have been performed if n (number of simulations) was equal to 0: the produced code would have stopped after the “parameters <- [. . . ]” line.
The second step consists of programming the simulations using the sets of previously coded parameter values. For the simulations, two additional tables are needed. First, a table containing the initial states of compartments is required. Initial states can be inputted directly as numeric values or formulas involving parameters (e.g., “kin/kout”). Initial states mentioned directly in the structural equations are automatically included in the pre-filled table. Second, a table must contain all information regarding one or several protocols of administrations (times of administrations, amount, inputted compartment, . . . ). Each protocol can contain one or several administrations. Of note, the time column can include any R code (text) which produces an atomic vector after evaluation (e.g., “0:10”, “seq(0,240,12)”, . . . ), allowing to create several administrations in a single line. Tlag, bioavailability and perfusions information are also set up in this table (in the Shiny app, new parameters corresponding to Tlag or bioavailability are automatically added to the parameters values table).
The simulations can then be performed using the deSolve package or the faster and more PMx-oriented RxODE package. Every possible combination of parameter values and protocols is simulated.
# Initial conditions for compartments states <- tibble(Cmt= c("Gut", "Central", "Periph"), t0 = 0) # Drug protocol definition (extension of deSolve events) events <- tribble(~Proto, ~var, ~time, ~value, ~method,~ADM, ~tlag, ~F, ~Perf, ~use, "1", "Gut", "0", 4.5 , "add","1" , F, F, "none", T, "2", "Gut", "seq(0,100,24)", 1 , "add","1" , F, F, "none", T ) # Function to add the RxODE expressions to paramInd simult <- make_simulations_rxode(parameter = paramInd,model = model_demo, states = states, events = events, times = c(seq(0,100,1)), Progress = F, returnExpr =T)
# Here we Print only the new expressions, as the # first nine ones correspond to paramInd expr({!!! simult[10:length(simult)]})
## {
## parameters_df <- ind_param %>% crossing(Proto = c("Prot 1",
## "Prot 2")) %>% rownames_to_column("id") %>% mutate(id = as.double(id))
## times <- c(seq(0, 100, 1))
## events_df <- tibble(Proto = c("1", "2"), cmt = c("Gut", "Gut"
## ), time = c("0", "seq(0,100,24)"), amt = c(4.5, 1), method = c("add",
## "add"), ADM = c("1", "1"), Perf = c("none", "none"), evid = c(1,
## 1)) %>% mutate(Proto = paste0("Prot ", Proto)) %>% mutate(time = map(time,
## ~eval(parse_expr(.x)))) %>% unnest(time) %>% bind_rows(crossing(time = c(seq(0,
## 100, 1)), evid = 0, cmt = "Gut", Proto = c("Prot 1",
## "Prot 2"))) %>% group_by(Proto) %>% nest() %>% full_join(parameters_df %>%
## select(id, Proto) %>% mutate(Proto = as.character(Proto))) %>%
## unnest() %>% mutate(amt = as.double(amt), time = as.double(time)) %>%
## arrange(id, time) %>% mutate(amt = as.double(amt), time = as.double(time)) %>%
## arrange(id, time)
## model <- RxODE({
## d/dt(Gut) <- -ka * Gut
## d/dt(Central) <- ka * Gut + Periph * k21 - Central *
## k12 - ke * Central
## d/dt(Periph) <- Central * k12 - Periph * k21
## conc <- Central/vd
## })
## simulations <- as_tibble(model$solve(parameters_df %>% select(-nameparset,
## -Proto), events_df)) %>% mutate(securejoin = 1) %>% left_join(parameters_df %>%
## mutate(securejoin = 1))
## }Not involved in the above example, tlag and bioavailability are applied directly by modifying the “events_df” table, respectively by an increase of the time of administration and by multiplying the amount of injected drug by the individual bioavailability. Perfusions are natively handled by RxODE package, while it was hard-coded (with an on/off time dependent zero-order input) when producing a deSolve code.
Regardless of the simulation software used (deSolve, RxODE), the simulated values can be plotted with two additional pieces of information: 1) a table containing which outputs the user wants to display and if observations points should be added (with various filtering systems); 2) a table containing for each plot (several can be produced simultaneously) additional information such as the scales to use (logarithmic or continuous) or the wrapping systems. It is possible to wrap the plot either by output, protocol, set of parameter values, a mix of them or none. In the following example, we wrapped the plot both by the set of parameter values and by protocol, every other characteristic (here the outputs) being dissociated using different colors (Figure 7).
# What to plot (output), and should we add observation points (not used here) plot_table <- tribble(~Plot, ~Todisplay, ~Point, ~Filter_of_dataset, ~YTYPE, ~Check, 1, "Periph", F, "", NA, T, 1, "conc", F, "", NA, T ) # Y logarithmic scale and grid “Parameters vs Events” (“P|E”) plot_table_cov <- tibble(Plot = 1, wrap = "P|E", ylog = T, xlog = F) # Function to add the plotting expressions to simult plotcode <- plot_simulations(simult, plot_table = plot_table, plot_table_cov = plot_table_cov, returnExpr = T) # Here also we print only the new expressions expr({!!! plotcode[15:20]}) ## { ## for (a in names(simulations)[!names(simulations) %in% c("id", ## "nameparset", "time", "Proto")]) { ## simulations[[a]] <- if_else(simulations[[a]] < 1e-04, ## 0, simulations[[a]]) ## } ## quantiles <- simulations %>% mutate(nameparset = fct_inorder(nameparset)) %>% ## gather(-id, -nameparset, -Proto, -time, key = "key", ## value = "value") %>% group_by(time, key, nameparset, ## Proto) %>% nest() %>% crossing(tibble(q1 = seq(0.1, 0.8, ## 0.1), q2 = q1 + 0.1)) %>% mutate(qx = map2_dbl(q1, data, ## function(q1, data) { ## unname(quantile(data$value, q1, na.rm = T)) ## })) %>% mutate(qx2 = map2_dbl(q2, data, function(q2, ## data) { ## unname(quantile(data$value, q2, na.rm = T)) ## })) %>% select(-data) ## breaks2 <- map(-40:40, ~1:9 * 10^.x) %>% reduce(c) ## labels2 <- as.character(breaks2) ## labels2[-seq(1, length(labels2), 9)] <- "" ## quantiles %>% mutate(color = key) %>% filter(key %in% c("Periph", ## "conc")) %>% mutate(forfct = as.double(nameparset)) %>% ## ggplot + geom_ribbon(aes(x = time, ymin = qx, ymax = qx2, ## alpha = paste0(q1, "-", q2), fill = fct_reorder(color, ## forfct))) + geom_line(data = quantiles %>% mutate(color = key) %>% ## filter(key %in% c("Periph", "conc"), q1 == 0.5), aes(time, ## qx, col = fct_reorder(color, as.double(nameparset)))) + ## labs(x = "Time", y = "", alpha = "quantiles", fill = "", ## col = "") + scale_alpha_manual(values = c(0.2, 0.4, ## 0.6, 0.8, 0.8, 0.6, 0.4, 0.2)) + theme_bw() + facet_grid(fct_reorder(nameparset, ## as.double(gsub(".+= *", "", nameparset))) ~ Proto) + ## scale_y_log10() ## }
# But we evaluate everything at the same time eval(expr({!!!plotcode}))
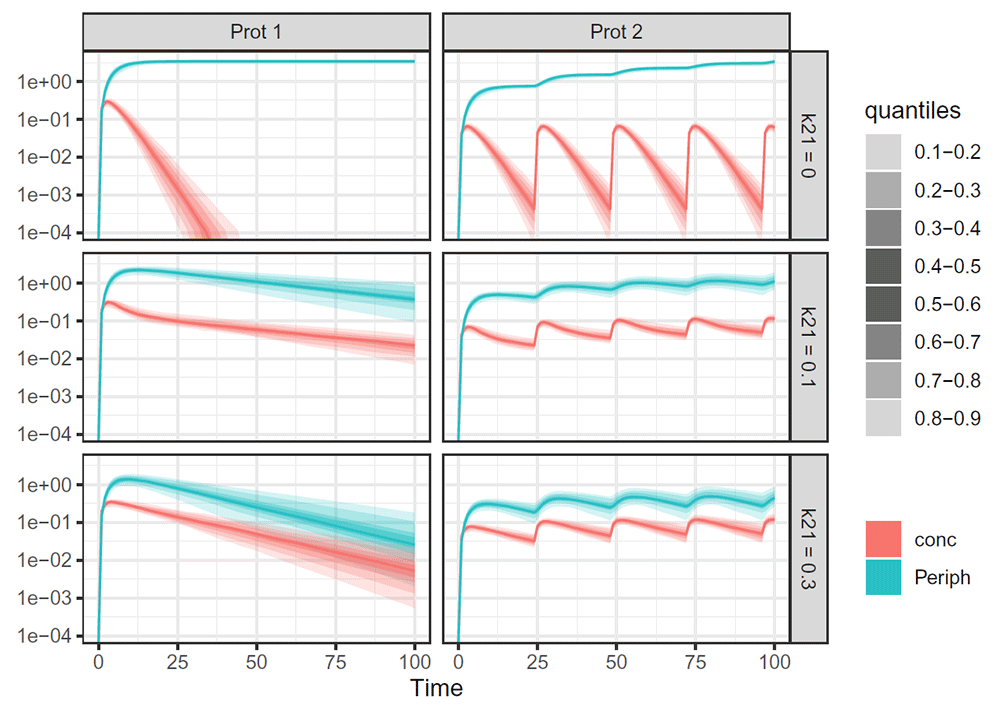
This plot corresponds to the evaluation of the final R expression, gathering both the parameter sampling, RxODE simulations, quantiles generation, and final ggplot pieces of codes.
Overall, this system is highly flexible with many different possible uses, such as exploring the dynamics of a model by looking at several variables simultaneously, exploring the impact of modifying parameter values, comparing different dose regimens,. . . Because it is possible to add observation plots (with various systems to control in which panels some subsets of data are displayed), different model structures or initial values can be tried for “manually” capturing the data before moving to a parameter estimation software. A recent feature allows having a first parameter estimation based on naive pool methods, using the R Optim function to optimize square residual sums.
Finally, Peccary can import and translate NONMEM, Monolix, or ADAPT files into simplified deSolve syntax. If the user provides a path to an estimated Monolix or NONMEM run, Peccary can extract the observation values, the estimated parameter values and the study designs for each individual (along with estimated typical parameters). Only the simplest study designs can be extracted for now. More complex input (e.g., NONMEM’s “SS” or “II” columns) are not covered yet and would require additional programming work. If the extraction is successful, all Peccary tables are automatically filled, with the possibility to switch between the population or any individual sets of values at any time. The main author particularly used this for first recreating the same individual profile as estimated by the parameter estimation software, before trying various modifications to explore what could improve the model performances (new structural equations and/or parameter values).
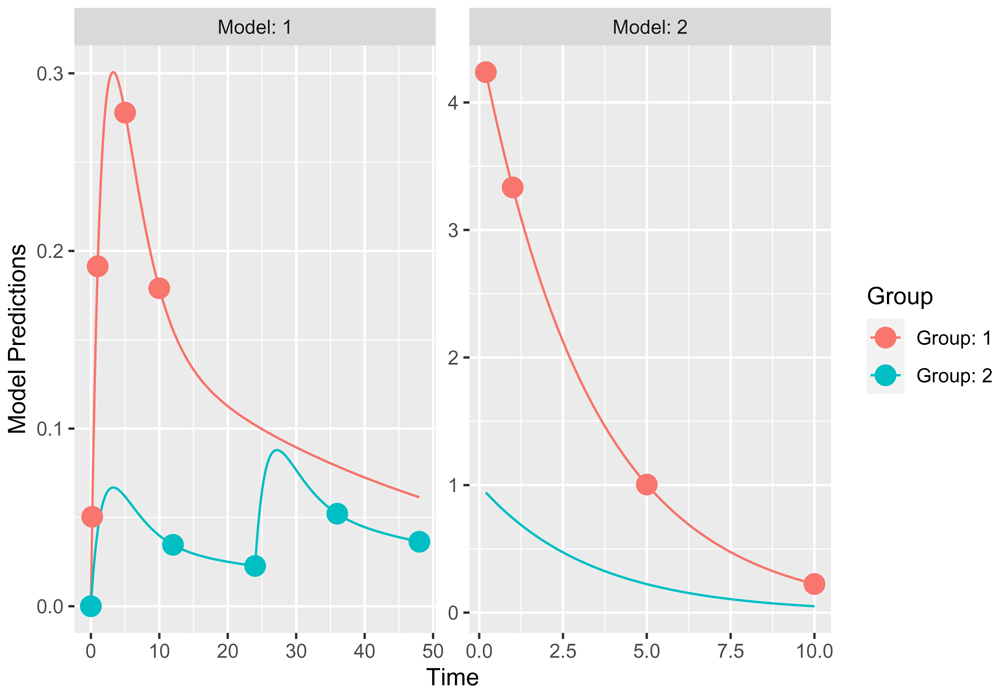
Design evaluation using PopED
PeccaReverse can also produce and evaluate a PopED code to perform design evaluation. The only additional required information is a table containing the desired sampling schedule per group (Figure 8). Several groups can be added and specific covariate values can be assigned to each. In the following example, two groups are created, respectively receiving protocols number one and two. The first group has two observation types (Gut and conc) while the second group has only one (conc). Two rows corresponding to the same group cannot have different protocols or numbers of ids, such as Non Available data (NA) are produced automatically in case of discrepancies (only the first values are kept). Similarly, each output can have a single set of error values (additive and/or proportional). A “F” after a number (in this table or in the variance/covariance matrix) stipulates that the corresponding value is fixed. The information of estimated or fixed typical parameter values is included in the parameter value table (column “E” of param_df ).
# To keep only one k21 values (vs three previously) param_df$Value <- c(0.3, 0.1,0.3,0.1,5) OD_input <- tribble(~Output, ~Group, ~Proto, ~ TimeSample, ~ add, ~ prop, ~nidgroup, ~cov, "conc", 1, 1, "c(0.2,1,5,10)", "0F", "0.2", 30, "", "Gut" , 1, NA, "c(0.2,1,5,10)", "0F", "0.1", NA, "", "conc", 2, 2, "seq(0,48,12)", NA, NA, 15, "") resPopEd <- pecc_PopEd(model = model_demo,OD_input = OD_input, states = states, events = events, parameters = param_df, diagOmega = matrix_sd, outputExpr = "Both") ## { ## library(PopED) ## modelpopEd <- function(Time, State, Pars) { ## with(as.list(c(State, Pars)), { ## dGut <- -ka * Gut ## dCentral <- ka * Gut + Periph * k21 - Central * k12 - ## ke * Central ## dPeriph <- Central * k12 - Periph * k21 ## conc <- Central/vd ## list(c(dGut, dCentral, dPeriph), c(conc = conc)) ## }) ## } ## events_input <- data.frame(var = c("Gut", "Gut", "Gut", "Gut", ## "Gut", "Gut"), time = c(0, 0, 24, 48, 72, 96), value = c(4.5, ## 1, 1, 1, 1, 1), method = c("add", "add", "add", "add", ## "add", "add"), Proto = c(1L, 2L, 2L, 2L, 2L, 2L), Group = c(1L, ## 2L, 2L, 2L, 2L, 2L)) ## model2 <- function(model_switch, xt, parameters, poped.db) { ## with(as.list(parameters), { ## A_ini <- c(Gut = 0, Central = 0, Periph = 0) ## times_xt <- drop(xt) ## eventdat <- events_input %>% filter(Group == group) ## times <- sort(c(times_xt, eventdat$time)) ## out <- ode(A_ini, times, modelpopEd, parameters, ## events = list(data = eventdat)) ## out <- out[match(times_xt, out[, "time"]), ] ## y <- xt * 0 ## y[model_switch == 1] <- out[, "conc"][model_switch == ## 1] ## y[model_switch == 2] <- out[, "Gut"][model_switch == ## 2] ## y = cbind(y) ## return(list(y = y, poped.db = poped.db)) ## }) ## } ## fg <- function(x, a, bpop, b, bocc) { ## parameters = c(ka = bpop[1] * exp(b[1]), ke = bpop[2] * ## exp(b[4]), k12 = bpop[3] * exp(b[3]), k21 = bpop[4] * ## exp(b[2]), vd = bpop[5] + b[5], group = a[1]) ## return(parameters) ## } ## feps_ODE_compiled <- function(model_switch, xt, parameters, ## epsi, poped.db) { ## MS <- model_switch ## y <- model2(model_switch, xt, parameters, poped.db)[[1]] ## Output1 <- y * (1 + epsi[, 1]) ## Output2 <- y * (1 + epsi[, 2]) ## y[MS == 1] <- Output1[MS == 1] ## y[MS == 2] <- Output2[MS == 2] ## return(list(y = y, poped.db = poped.db)) ## } ## poped.db <- create.poped.database(ff_fun = "model2", fError_fun = "feps_ODE_compiled", ## fg_fun = "fg", groupsize = c(30, 15), m = 2L, sigma = c(conc_prop = 0.2, ## Gut_prop = 0.1), notfixed_sigma = c(conc_prop = 1, ## Gut_prop = 1), bpop = c(ka = 0.3, ke = 0.1, k12 = 0.3, ## k21 = 0.1, vd = 5), notfixed_bpop = c(1, 1, 1, 1, ## 1), d = c(ka = 0.3, k21 = 0.3, k12 = 0.3, ke = 0.3, ## vd = 0.3), notfixed_d = c(ka = 1, k21 = 1, k12 = 1, ## ke = 1, vd = 1), xt = list(c(c(0.2, 1, 5, 10), c(0.2, ## 1, 5, 10)), seq(0, 48, 12)), model_switch = list(c(1, ## 1, 1, 1, 2, 2, 2, 2), c(1, 1, 1, 1, 1)), a = list(group = 1, ## group = 2)) ## popedResult <- list() ## popedResult$plot_OD <- plot_model_prediction(poped.db, model_num_points = 500, ## facet_scales = "free") ## popedResult$result_OD <- evaluate_design(poped.db) ## names(popedResult$result_OD$rse) <- c("ka_pop", "ke_pop", ## "k12_pop", "k21_pop", "vd_pop", "ka_omega", "ke_omega", ## "k12_omega", "k21_omega", "vd_omega", "conc_prop", "Gut_prop") ## popedResult ## } # Print the predicted Relative Standard Errors (RSE) resPopEd$result_OD$rse ## ka_pop ke_pop k12_pop k21_pop vd_pop ka_omega ke_omega ## 10.085469 32.911456 26.134697 48.565118 7.533477 26.525564 187.375865 ## k12_omega k21_omega vd_omega conc_prop Gut_prop ## 79.797228 89.814517 229.365823 13.630363 14.894043
# Print the study design plot resPopEd$plot_OD
Translation into other syntaxes
The model can also be translated into various other syntaxes such as Monolix, NONMEM, ADAPT, and nlmixr. All the corresponding translation functions are based on regular expression manipulation (mostly using gsub function) and take into consideration the information included in the previous tables (e.g., parameter distribution, initial parameter values, . . . ). A final table can also mention the outputs (with associated YTYPE and error model). For example, the following code translates our demonstrative model into a Monolix model file.
mb_output <- tibble(output = "conc", YTYPE = 1, err_add = 0.1, err_prop = 0.3, export = TRUE, rm = FALSE) ode_monolix(model_demo, parms = param_df, y = states, mb_output = mb_output, depot = events) ## [LONGITUDINAL] ## input ={ka, ke, k12, k21, vd} ## PK: ## depot(target = Gut, adm = 1) ## EQUATION: ## ; Initial conditions ## t0 = 0 ## Gut_0 = 0 ## Central_0 = 0 ## Periph_0 = 0 ## ; Dynamical model ## ddt_Central = ka * Gut + Periph * k21 - Central * k12 - ke * Central ## ddt_Periph = Central * k12 - Periph * k21 ## conc = Central / vd ## ## ## OUTPUT: ## output = {conc}
A function for creating the corresponding mlxtran file (to use directly through R with the Lixoft connector32) exists and works for the simplest models (e.g., one or several YTYPE with normal or log-normal variabilities but without covariate), but was in practice judged not efficient enough compared to simply copy/pasting the structural model code directly in the Monolix suite, and thus not further developed.
For NONMEM, minor manual modifications would be required after the translation, for instance if the user wants to change the used ADVAN or the estimation method. Parameters can be coded with or without MU referencing, and a block for handling data below the levels of quantification can be automatically added.
ode_nonmem(model_demo,omega = matrix_sd, parms = param_df, y = states, mb_output = mb_output, mu_referencig = F) ## $PROB name of the model and/or description ## ## $INPUT ## ## $DATA your_file.data IGNORE = I ## ## $SUBROUTINE ADVAN13 TOL9 ## ## $MODEL ## COMP = (Gut) ## COMP = (Central) ## COMP = (Periph) ## ## $PK ## ## ka = THETA(1) * exp(ETA(1)) ## ke = THETA(2) * exp(ETA(2)) ## k12 = THETA(3) * exp(ETA(3)) ## k21 = THETA(4) * exp(ETA(4)) ## vd = THETA(5) + ETA(5) ## ## A_0(1) = 0 ; Gut initialisation ## A_0(2) = 0 ; Central initialisation ## A_0(3) = 0 ; Periph initialisation ## ## $DES ## ## DADT(1) = -ka * A(1) ## DADT(2) = ka * A(1) + A(3) * k21 - A(2) * k12 - ke * A(2) ## DADT(3) = A(2) * k12 - A(3) * k21 ## conc = A(2) / vd ## ## $ERROR ## ADD = THETA(6) ## PROP = THETA(7) ## OUTPUT =conc ## IPRED = OUTPUT ## W = (OUTPUT * OUTPUT * PROP**2 + ADD**2)**0.5 + 0.0001 ## IRES = DV - IPRED ## IWRES = (DV - IPRED)/(W) ## ## Y = OUTPUT + W * EPS(1) ## ## $THETA ## (0,0.3) ; ka - theta 1 ## (0,0.1) ; ke - theta 2 ## (0,0.3) ; k12 - theta 3 ## (0,0.1) ; k21 - theta 4 ## (0,5) ; vd - theta 5 ## (0,0.1) ; err_add - theta 6 ## (0,0.3) ; err_prop - theta 7 ## ## $OMEGA ## 0.3 ; ka - eta 1 ## 0.3 ; ke - eta 2 ## 0.3 ; k12 - eta 3 ## 0.3 ; k21 - eta 4 ## 0.3 ; vd - eta 5 ## ## $SIGMA 1 FIX ## ## $COV MATRIX=S ## ## $EST METHOD=SAEM LAPLACIAN INTERACTION NBURN = 1000 NITER = 300 ISAMPLE = 2 SIGL = 10 CTYPE = 3 ## ## $TABLE ID TIME DV IPRED ONEHEADER NOAPPEND NOPRINT FILE = youroutputfile.TAB
PeccaResult
Various software tools allow the estimation of population parameters, whether they are proprietary (NONMEM, Monolix, ADAPT) or open-source (Saemix, nlmixr), producing similar outputs (parameter estimates, objective function, simulated values, . . . ), but often stored with different architecture. The goal of PeccaResult is thus to provide a tool to easily compare various runs together independently of the parameter estimation software by first standardizing the output of each software in a uniformized structure before manipulating those structures with new model diagnostic functions. However, metaprogramming such uniformization of software is very challenging and would probably result in a final code far too complex compared to the direct use of other specific software (e.g., Monolix Suite or Xpose for NONMEM runs23). Therefore, PeccaResult was designed to be used only during the modeling process (the trial-and-error phase), and not for being integrated into a final report: its integration into a traceable workflow is currently not available.
The main idea of PeccAnalysis is thus to use various functions to standardize the output of each software and store their data into a unique structure, namely a “run” S4 object38. This object contains essentially a list of various tables: one for estimates of population parameters, one for the individual parameters obtained through posthoc analysis, for the individual and population predictions, etc. By giving a path to a run (a NONMEM or ADAPT result file, a Monolix output folder. . . ), a function automatically recognizes the software and performs this run standardization. Each software requires unique steps for extracting the possible information. More information is available on GitHub under the corresponding source code. Each run can be created from scratch or from a second S4 object, called “folder”, containing several runs. Concretely, the user must use a specific closure function39 that takes the path of any folder :
demo <- createFolder("D:/.../anyfolder")Several internal steps occur using createFolder() function. First, an algorithm searches for every run (for now Monolix 2016 and similar, Monolix 2019 and similar, NONMEM, ADAPT and nlmixr) contained in this folder or any subfolders. It produces a table containing one run per row, with its complete path, source software, objective function value and an ID number (Figure 9A). Second, this table is injected in an S4 folder object, containing an initially empty list used later to store desired runs. Third, a function is returned to manipulate this enclosed folder object. This function takes as an argument the ID number(s) of desired runs:
demo(1) # This returns the run object of run number 1
demo(1:2) # This returns the folder object containing run number 1 and 2The first time a run is requested, its creation (which takes a few seconds) is directly followed by its storage inside the “folder” object. The next time this run is requested, its creation is not re-performed.
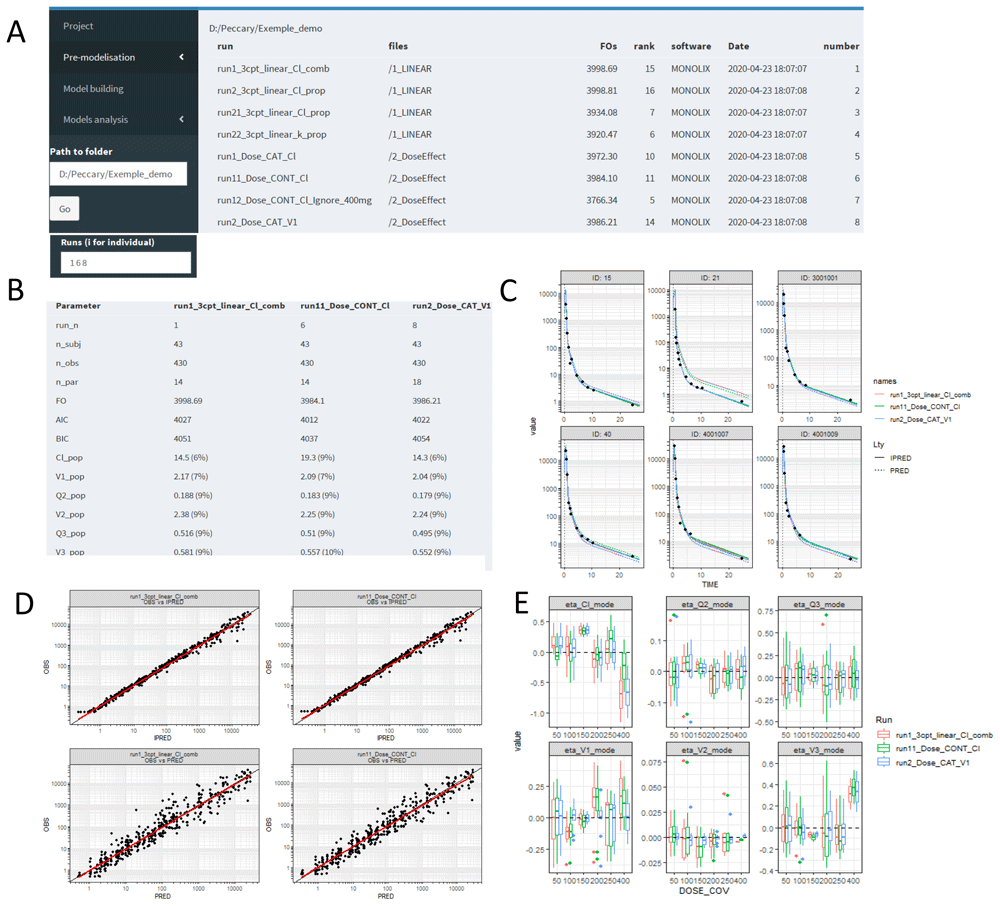
A) The user provides a folder path (left dark panel), clicks on “go”, and a table is created (right) containing basic information and a unique ID for every runs found on the root folder and its subfolders. B) By selecting desired run IDs (here, 1, 6 and 8), a comparative table is created with one run per column and one result value per row. C) Individual (straight lines) and populational (dashed lines) fits with runs comparisons (one color per run). D) Sample of two Goodness of fit plots (one per row) for two runs (one per column). E) Covariate analysis (here, the dose considered as categorical) with runs comparison (one color per run).
Various functions have then been created to analyze either a single run (if a “run” object is passed as an argument), or to compare runs (if a “folder” is inputted), for instance:
results(demo(1)) # return the result of run number 1
results(demo(c(1,3))) # compare the results of run 1 and 3This results() function provides the main results of a model, namely the criteria score (objective function, AIC, BIC), the parameter estimates with precision and, if provided, the shrinkage. If several run IDs are entered, a full join is automatically created to compare the run values (Figure 9B). An individual prediction (IPRED) function plots IPRED and PRED vs. time for each individual and each YTYPE. If several run IDs are inputted, the predictions will be plotted in the same graph with different colors (Figure 9C). The goodness of fit plots can be stratified by covariate, and users can specifically compare any subplot between desired runs (Figure 9D). The same system applies for random effect analysis (variance-covariance plots) or covariate analysis (Figure 9E). vpc25 and coveffectsplot40 packages have also been implemented in the Shiny app to simplify the production of visual predictive checks and forest plots, respectively.
The strength of this system is the universality of these functions. If one creates a new function working with runs, this function will directly work with all software (NONMEM, Monolix. . . ). Reciprocally, if one adds a new software standardization (e.g., from Phoenix WinNonLin), then all Peccary functions will be directly available. The currently available functions cover the most common analysis performed after a run estimate. However, more complex projects often require very specific outputs. Users can then manually create a function using standardized run and import them into a project. Of final note, some parameter estimation software, for instance NONMEM, allows syntax flexibility, creating “personal ways of coding”. Those syntax variabilities can create difficulties for PeccaResult to extract all the needed information effectively. If previous effort has been made to consider several “personal ways” of coding in NONMEM, more work is required to make those extractions as flexible and adaptive as possible. This challenge and the difficulty of metaprogramming the system explain why PeccaResult should be used with caution and not integrated into a report. It remains, nevertheless, very useful to perform quick “informal” run results, visualization, and run comparisons.
The use of multiple software is essential for pharmacometricians to improve efficiency, as no single program can perform all desired analyses. However, this creates significant efficiency issues. In this context, Peccary is an R project created with the sole objective of improving the efficiency of pharmacometricians. So far, the most complex model on which Peccary has been used in real conditions (using both PeccAnalysis, PeccaReverse and PeccaResult functions) was a Monolix PK/PD model containing complex if-else structures, multiple covariates, 51 parameters, 18 ODEs, and five observation types fitted simultaneously.
From many aspects, Peccary tried to reproduce the markdown pandoc efficiency but applied in PMx. First, Peccary can accelerate the use of various R packages (pknca, RxODE, PopED, table1,. . . ) by simplifying their syntax, and hence reducing 1) the required knowledge and 2) the time needed before launching these analyses. Second, Peccary can create links between the different packages. For instance, the same input tables in PeccaReverse are used both for simulations, design evaluation and syntax translations, improving the efficiency at the multi-software (or interoperability) level. The Shiny application is fast enough to be used during a live meeting, dynamically producing plots or analyses. Similarly, the Shiny app has educational potential, especially with its ability to easily reload any dataset or model simulations. In a second time, and thanks to its metaprogramming systems, it is possible to extract the peccary independent R code from most analyses, before copy/pasting it into a report to be checked and, if needed, secondarily modified. It is almost always faster to create a code through Peccary than coding it from scratch, even if secondary modifications are needed. If knowing the pre-existing packages integrated in Peccary is not mandatory to produce many outputs, it is nevertheless useful to learn them in order to reach the full potency of the system.
The Peccary project started in mid-2018 as a personal side project supporting the modeling work of the lead author, with no release ambition. After four years of adding different features, the current version of Peccary significantly improves the efficiency of PMx on a daily basis. Consequently, Peccary may be of interest to other pharmacometricians and is now aiming to be released. However, scaling such a project from a single user to multiple people is challenging. The chances of success would be optimized if the project became fully collaborative, in line with bazaar development. Thanks to its R-only structure and high modularity, every user can become a co-developer by checking, improving, or adding new features to Peccary. Being frequently updated, each modification or addition of a feature is registered in the “author section” in the Shiny app (authors listed by alphabetic order). Finally, any part of Peccary can be integrated in any other project without the authors’ initial authorization.
Due to the bazaar development approach of Peccary, there is a large heterogeneity in terms of Peccary function qualities. Some of them have been tested, used, improved and fixed several times until complete stability. Some others (the most recent or the less used ones) are more likely to contain errors or to be incomplete. Nevertheless, Peccary was designed to be useful even if imperfect and unvalidated. Despite the integration of several unit tests (e.g., for the peccary_nca() function, more information on GitHub), it is always the user’s responsibility to verify the results, as the purpose of Peccary is only to speed up the creation of R codes and have quick access to various results. Users can verify these outputs in two ways, 1) via the metaprogramming system and 2) by comparing the Peccary result to any other software making the same result. For the metaprogrammed functions, computational errors can occur either during the internal Peccary metaprogramming task or during the code evaluation. In the last case, it is still possible to extract the metaprogrammed code and manually fix it (most of the time requiring little effort and time). Above all, multiplying the numbers of co-developer would accelerate the detection and correction of these bugs, in accordance with Linus’s law “given enough eyeballs, all bugs are shallow”33.
There are also many features that need to be added. For example, the current version of Peccary does not support Time-To-Event data, mixture model, Bayesian analysis, and the Phoenix WinNonLin translation tool sets. . . . It should be noted that other types of ODE-based model types (QSP or PBPK modeling for instance) can be imported or created inside PeccaReverse. Being able to launch and program run parameter estimations through an R package (nlmixr, saemix) or external software (NONMEM, Monolix. . . ) within Peccary can also be of great interest, requiring appropriate parallelization and computational server communications. Finally, any R package can potentially be metaprogrammed in Peccary to further merge more open-source tools within the same platform.
In summary, Peccary is an R package that increases the daily efficiency of a pharmacometrician. Although Peccary was initially developed as an individual project, it has now grown into a platform that aims to benefit the workflow process development for all pharmacometricians by integrating the different steps of the PMx into one tool. However, in line with the bazaar model, Peccary will only reach its full potential through a fully collaborative process.
Peccary source code:
Software available using the following R code (automatically downloading all the subpackages): devtools::install_github("Peccary-PMX/Peccary")
Source code available from: https://github.com/Peccary-PMX/Peccary
Archived source code at time of publication: https://doi.org/10.5281/zenodo.684180141
License: GNU General Public License v3.0.
PeccAnalysis source code:
Software available using the following R code (already downloaded with Peccary main package): devtools::install_github("Peccary-PMX/PeccAnalysis")
Source code available from: https://github.com/Peccary-PMX/PeccAnalysis
Archived source code at time of publication: https://doi.org/10.5281/zenodo.684182342
License: GNU General Public License v3.0.
PeccaReverse source code:
Software available using the following R code (already downloaded with Peccary main package): devtools::install_github("Peccary-PMX/PeccaReverse")
Source code available from: https://github.com/Peccary-PMX/PeccaReverse
Archived source code at time of publication: https://doi.org/10.5281/zenodo.684182943
License: GNU General Public License v3.0.
PeccaResult source code:
Software available using the following R code (already downloaded with Peccary main package): devtools::install_github("Peccary-PMX/PeccaResult")
Source code available from: https://github.com/Peccary-PMX/PeccaResult
Archived source code at time of publication: https://doi.org/10.5281/zenodo.684183344
License: GNU General Public License v3.0.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)