Keywords
cell senescence, RNA-seq, quantitative proteomics, integrative analysis, U2OS
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Cellular Senescence in Stress and Aging collection.
cell senescence, RNA-seq, quantitative proteomics, integrative analysis, U2OS
Cellular senescence describes the irreversible loss of cells’ ability to proliferate. This phenomenon was first described by Hayflick and colleagues six decades ago when they showed that cultured human fibroblast cells have a limited capacity to proliferate.1 This durable cell cycle arrest is resistant to mitogenic stimuli and distinct from other hyporeplicative states such as quiescence or terminal differentiation. First considered an artefact of in vitro cell culture, senescence is now considered a fundamental cellular process associated with a broad range of developmental and pathological processes and an irrefutable hallmark of organismal ageing.2,3
Replicative senescence is induced through telomere attrition; however, senescence may also be induced by several other intrinsic stressors such as oxidative stress, oncogene activation, or genomic instability.4 Senescence is also induced through extrinsic stimuli including viral infection, radiation, and chemotherapeutics.4,5 The onset of senescence is associated with a range of molecular and morphological traits including the expression of several senescence markers and a significantly enlarged and flattened appearance.6,7 The most commonly employed method to identify senescent cells is to stain for senescence-associated β-galactosidase activity (SA-β-gal). This assay exploits the unique pH of senescent cell lysosomes (pH 6.0), which can be detected using X-gal staining. Additional molecular characteristics include the formation of senescence-associated heterochromatin foci (SAHF) and the activation of p53 and p21, occurring at the onset of cell cycle arrest, which is subsequently maintained by the constitutive activation of p16.8,9
Cellular senescence is classically considered an anti-tumour mechanism, acting as a barrier to proliferation in the event of significant cellular damage.10 Although unable to divide, senescent cells remain metabolically active allowing them to participate in a range of physiological processes and secrete a range of potent inflammatory proteins known as the senescence-associated secretory phenotype (SASP).11 The SASP is thought to have evolved to aid in eliminating senescent/neoplastic populations through the recruitment of immune cells.3,12 However, as the number of senescent cells increases with age, the constant production of these inflammatory proteins promotes tissue dysfunction and contributes to a range of age-related diseases such as cardiovascular disease, diabetes, and cancer.11,13–15 Thus, senescence is highly pleiotropic and exists as part of a complex balance to maintain the function and health of cells, tissues, and organisms.
Senescence and apoptosis are closely linked and together form the primary protective mechanisms to suppress tumorigenic events. Dysregulation in the apoptotic apparatus is well described during neoplastic transformation, and there is increasing evidence that pathways of senescence induction are also inhibited.16 Several chemotherapeutic drugs have been shown to induce senescence in cancer cells. An example is the topoisomerase inhibitor doxorubicin, used clinically to treat cancers of the blood, stomach, lungs, and ovaries (amongst others), which disrupts the re-ligation of DNA strands and leads to the activation of the DNA-damage response.17–19 Although inducing a non-proliferate state in tumour cells may appear to be a favourable outcome, it is likely to be a heterogeneous response with one population entering complete senescence whilst others continue to proliferate.16 Moreover, the induction of senescence and subsequent secretome produce a microenvironment conducive to tumourigenesis and/or disease relapse.16 It is also possible that this heterogeny gives rise to a more aggressive tumour population.
Understanding the mechanisms through which cancer cells can escape proliferative arrest, which is otherwise expected in surrounding normal cells, is essential to understanding how malignant cells resist genotoxic drug therapy.16 Here we have integrated RNA-Seq and proteomic analyses to investigate the transcriptome of a model of chemotherapy-induced senescence in a common cancer cell line. We have induced senescence using doxorubicin, tracked the development of senescence and compared our data to existing data in commonly used models of cellular senescence.
Wild type U2OS cells were a kind gift from Dr Nancy Kedersha, Harvard medical school, USA.20 Cells were maintained at 37°C, 5.0% (v/v) CO2, and 95% humidity and passaged when they reached 90% confluency. All cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% foetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 ug/ml streptomycin.20
Senescence was induced via incubation in 200 nM doxorubicin for 48 hours. The media was then exchanged and the cells cultured for an additional 5–7 days. Senescent cells were identified via staining with senescence associated -galactosidase (SA--Gal) staining solution (150 mM NaCl, 200 mM MgCl2, 40 mM citric acid, 12 mM sodium phosphate, 5 mM potassium ferrocyanide and 5 mM potassium ferricyanide, adjusted to pH 6.4). Cells were fixed in 4% PFA prior to incubation with the staining solution overnight at 37°C. Cells were imaged using a bright field Evos XL Core Cell Imaging microscope.
Cells were washed in PBS and lysed in RIPA buffer (150 mM NaCl, 1 percent Nonidet P-40, 0.1 percent SDS, 0.1 percent sodium deoxycholate, 50 mM Tris (pH 7.4)) and centrifuged at 20,000g for 20 min at 4°C. The protein concentration of the supernatant was measured using Pierce™ BCA assay kit (ThermoFisher Scientific, 23225). Preparation of peptide samples for proteomic analysis and mass spectrometry was performed by the proteomics facility at Durham University Biosciences as described before21 using a FASP Protein Digestion Kit (Expedeon 44250) and sequencing grade-modified trypsin (Promega, V5111). Spin-filter eluates were de-salted using C18 ZipTips (Millipore) following freeze-drying and resuspension in 3 percent acetonitrile, 0.1 percent TFA. Each sample fraction analyzed contained 5g peptides. Samples were loaded and washed on a TriArt C18 Capillary guard column 1/32”, 5m, 50.5 mm trap column (YMC) and online chromatographic separation performed over 57 min on a Triart C18 Capillary column 1/32”, 12 nm, S-3 m, 150 0.3 mm (YMC) at a flow rate of 5 l/min with a linear gradient of 3–32 percent acetonitrile, 0.1 percent formic acid over 43 min, then to 80 percent acetonitrile, 0.1 percent formic acid over 2 min, held for 3 min before returning to 3 percent acetonitrile, 0.1 percent formic acid and re-equilibrated. Analysis was carried out on an Ekspert™ nanoLC 425 with low micro gradient flow module (Eksigent), coupled to a quadrupole Time-Of-Flight (QTOF) mass spectrometer (TripleTOF 6600, SCIEX, MA) with a DuoSpray source (SCIEX) and a 50-micron ESI electrode (Eksigent). SWATH acquisition was for 55 min with a 3.2 s cycle time. Each cycle consisted of MS-spectrum acquisition at 400 to 1,250 m/z for 250 msec followed by MS/MS (100 to 1500 m/z) using 100 variable SWATH windows (parameters downloaded from22), 25 msec accumulation for each in high sensitivity mode with rolling CE and 2+ ions selected. Samples were spiked with iRT peptides (Biognosys) at a ratio of 1g protein to 0.1l 10 RT peptide mix. The data acquired from each scan cycle (400–1250 m/z) was processed using SCIEX version 1.7.1 software.23 Three biological replicates for each condition (young vs senescent) were prepared for analysis. For each biological replicate, three technical replicate LCMS runs were undertaken. This resulted in nine total replicates for each condition.
PeacView 2.224 was used to obtain raw counts of peptide distribution which were normalized on peak areas using MarkerView 1.2.25 To calculate the fold change between young vs. senescent cells, a t-test of the nine senescent cell samples against the nine young samples followed by a two-sample t-test for each experiment and per gene was carried out. This output contains fold changes as well as p-values. The p-adjusted values were then calculated using the Benjamini-Hochberg procedure in R (package stats (version 3.6.2), R version 4.1.226).
Statistical testing for overrepresentation or enrichment of REACTOME terms was performed using the R package Reactome Pathway Analysis version 1.38.027 with the conditions pvalueCutoff = 0.05, pAdjustMethod = “BH”, qvalueCutoff = 0.2, minGSSize = 10 and maxGSSize = 500.
Cells were then fixed in 4% paraformaldehyde for 15 min at room temperature prior to permeabilisation in 0.5 pc Triton X-100 for 20 min. Cells were incubated in blocking buffer (3 pc BSA in PBS) for a minimum of 30 min. Primary (SAHF, 1:500 (Proteintech catalog number AB8898) and secondary antibodies (Cy3 conjugated anti-rabbit (Jackson Immunoresearch, catalog number 711-165-152) were diluted in blocking solution (3% BSA in PBS) and incubated with the cells for at least one hour. Cells were stained with DAPI staining solution for 15 min (40 ng/ml DAPI in PBS) and the coverslips were subsequently mounted with Vectashield mounting media (Vector Labs catalog number H-1900). Imaging was performed using a Zeiss 880 line scanning confocal microscope.
Total RNA was isolated from young and senescent U2OS cell cultured using Trizol reagent (Sigma, T9424). Cells were seeded at per well in 6 well tissue culture plates and harvested in 300 l Trizol after washing twice in PBS. Samples were incubated for 5 minutes at room temperature. 200 l chloroform was the added to each sample, incubated for 5 minutes at room temperature and then centrifuged at 12,000g for 15 minutes at 4 degrees Celsius. The upper aqueous phase was carefully removed. RNA was precipitated via the addition of 250 l isopropanol and pelleted via centrifugation at 12,000g for 10 minutes at 4 degrees Celsius. The pellet was washed twice in 70pc ethanol, air dried, and re-suspended in RNAse-free water. RNA yield and purity was determined using a nanodrop spectophotometer (ThermoScientific ND-1000). Library preparation and sequencing was performed at the DBS-Genomics sequencing facility, Durham University. Extracted RNA was further purified using DNAse TURBO and RNA purity and concentration was evaluated on TapeStation (Agilent Technologies). Library preparation was undertaken using 1 microgram purified RNA using the NEBNext rRNA Depletion Kit (Human/Mouse/Rat) (E6310) followed by the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760). The indexes were also from NEB, NEBNext Multiplex Oligos for Illumina (Set 1, E7335). Libraries thus prepared were run on an Illumina NextSeq 4500 at 100 cycles 100bp paired end module. The quality of the raw data was controlled with FastQC 0.11.9,28 reads found to be sub-optimal were removed from the analysis pool, for example adapters and bases with an overall quality below 15 in a sliding window of size 4bp were trimmed with Trimmomatic 0.38.29 Annotation of the data to the human genome (GRCh38) was carried out using STAR 2.7.0f.30 Fold changes with corresponding p-adjusted values were calculated using DESeq2.31
WT U2OS cells were harvested on Day 0 (when Doxorubicin was added), Day 2 (when doxorubicin was removed), Day 5, Day 7 and Day 9 prior to staining with SA--Gal solution. The blue colouration is indicative of a senescent phenotype with the intensity increasing with time after dox treatment. The first senescent cells were detected at Day 2 and the increased intensity of the stain indicated that most cells were senescent by Day 7 (see Figure 1a). At Day 7 the cells showed a significant increase in SA--Gal staining (fold change of four compared to day 5). The percentage of SA--Gal positive cells was analysed using ImageJ32 with version number 2.0.0-rc-69/1.52p. By Day 9 a decline of 1.25-fold in the SA--Gal positive cells was detected compared to Day 7.
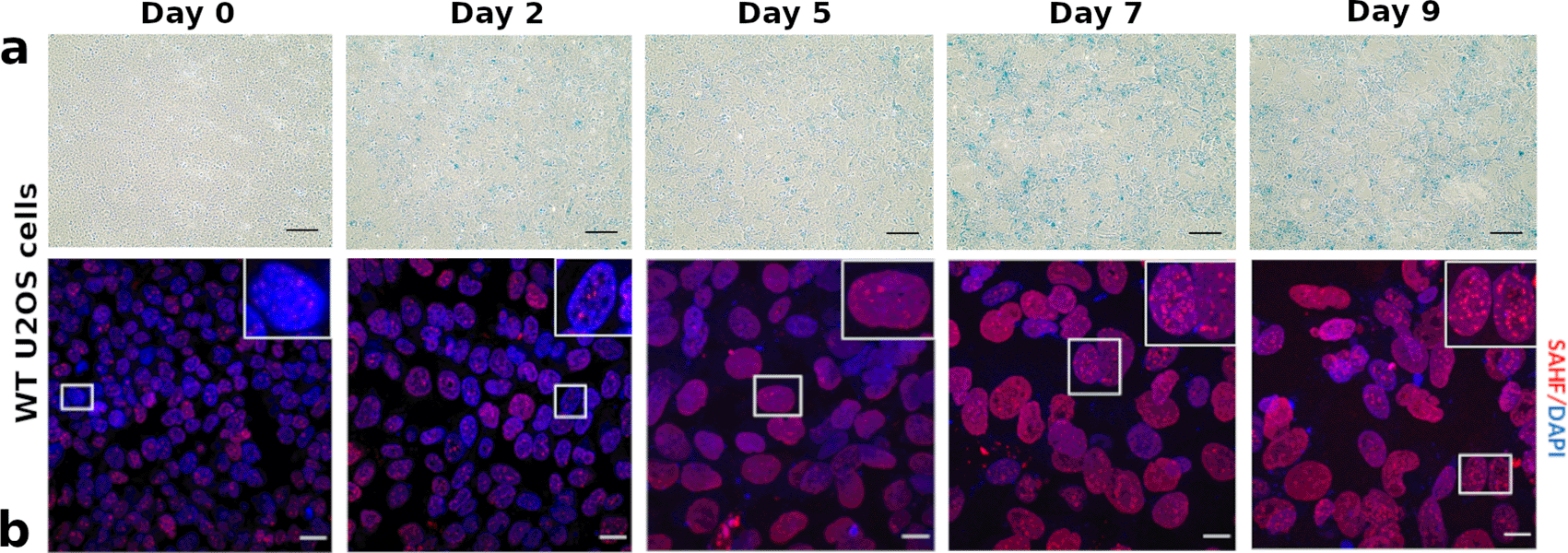
(a) SA--Gal positive cells were detected in WT U2OS cells. Cell senescence was induced using 200 mM doxorubicin. Cells were fixed and incubated with SA--Gal staining solution overnight at 37°C. Blue stains were detected in WT U2OS cells at Day 2 post-doxorubicin treatment. Gradual increase in the intensity of blue stain was observed. The blue stain indicates SA--Gal positive (senescent) cells. Scale bar = 100. (b) WT U2OS cells were treated with 200 mM doxorubicin for 48 hours. Cells were collected at Day 0, 2, 5, 7, 9, fixed and stained with SAHF antibody (red). WT U2OS cells showed no/few SAHF (less than 5 foci per cell) at Day 0, 2 and 5. On Day 7 and 9, they showed an increased number of SAHF. Scale bar = 20 um.
We employed a second method to assess the senescence induction protocol by measuring senescence-associated heterochromatin foci (SAHF). SAHF are formed when the chromatin in the nucleus of senescent cells undergoes remodeling by forming domains of heterochromatin.33 SAHF formation in U2OS cells was examined following the treatment routine with Doxorubicin. This was followed by immunofluorescence microscopy using a histone H3 (tri methyl K9) antibody that targets the nucleosome and provides a widely used read-out for SAHF. SAHF formation was monitored on Day 0, 2, 5, 7, and 9 post-doxorubicin treatment in Figure 1b. Cells that are forming five or more SAHF are counted and considered senescent. WT U2OS cells showed fewer than five structures on Day 0, 2 and 5. On days 7 and 9 the number of SAHF increased and were quantitatively analysed using ImageJ. At Day 7, cells showed a 3.75-fold change increase in the number of SAHF-forming cells of compared to Day 5 cell cultures and a further increase of 2.5-fold change at Day 9 compared to Day 7. Indeed, by Day 9, approximately 47% of U2OS cells were detected to form more than five SAHF foci per cell.
A total of 5335 proteins were quantified in our measurements, these are listed in Table S1 in Ref. 34. Of these 2672 had a p-adjusted value below 0.05, and are visualized in Figure 2 as a volcano plot. Data points in grey represent genes that show less than two-fold differential expression, i.e. have a and are therefore not significantly different between proliferating and senescent cells. Genes with a regulation between are significantly expressed and are depicted as small black dots. Highly differentially expressed genes () are indicated by large black markers and are also labeled with their gene name. We found 211 up-regulated () proteins of which 17 were highly up regulated (). Interestingly, only 91 proteins were downregulated (), among these were six which were strongly downregulated (). A list of the highly expressed proteins can be found in Supplementary Table S2 in Ref. 34.
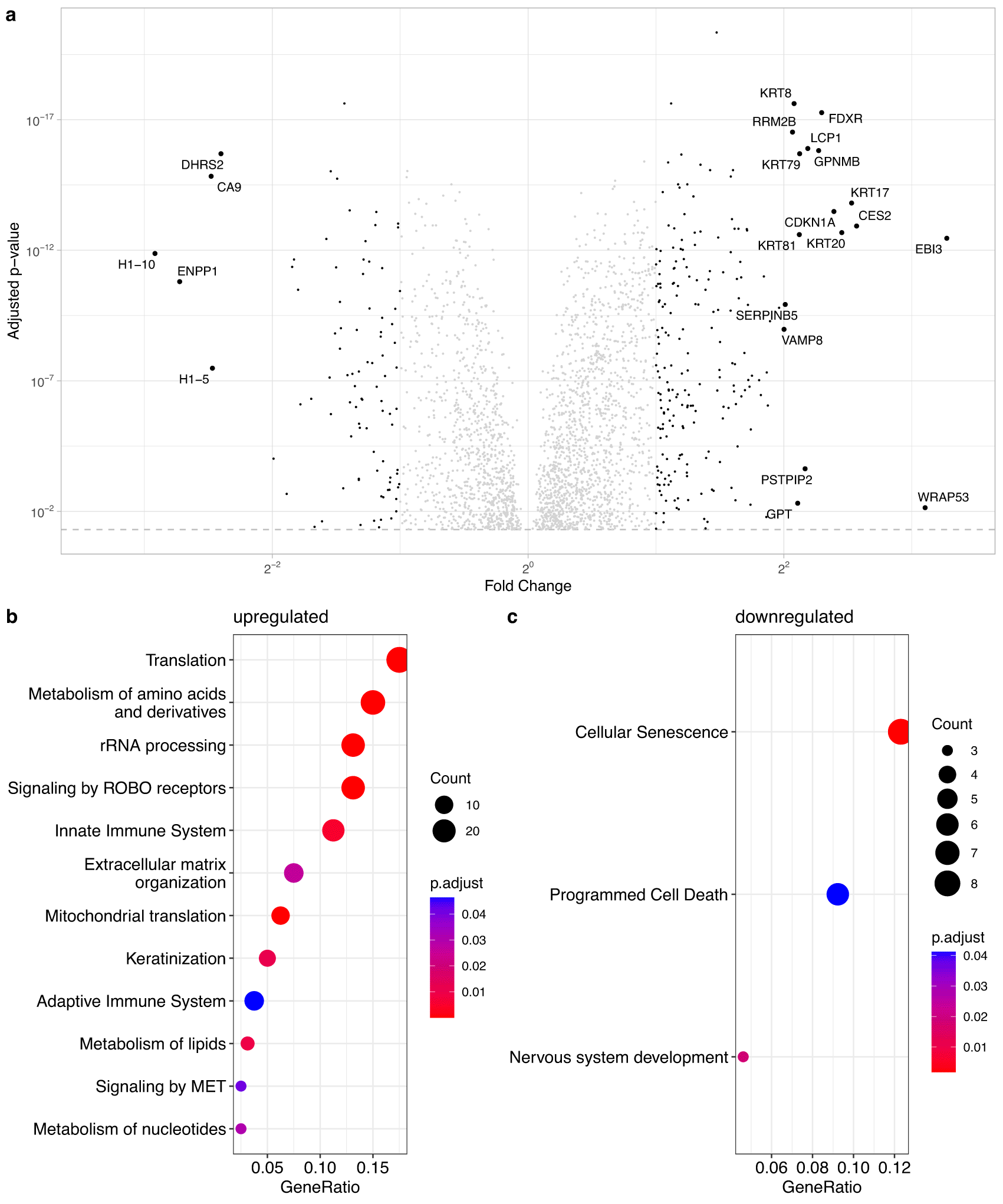
(a) Volcano-plot for all U2OS proteins with significant p-adjusted value (p-adjusted value >0.05). Proteins with a are not significantly regulated and shown in grey; expressed proteins are significantly regulated (1 < < 2) and marked with small black points. Highly expressed proteins ( > 2) are shown with big dots and are labeled with the gene name. (b and c) Identification of significant over-represented/underrepresented processes in senescence. Part a shows subsumption of Reactome terms of importance for the upregulated proteins (). Part c shows the significantly underrepresented processes in senescence (underlying proteins have a ).
The list of significantly enriched proteins (, p-adjusted value below 0.05) were investigated with Reactome35 to uncover functional pathways impacted during senescence. Figure 2b and c show the pathways, which significantly change in response to senescence induced with doxorubicin. Due to the large numbers of pathway hits, we organised the results such that the parent pathway is emphasised and child pathways that share an enriched parent pathway are ignored. Furthermore, two or more child pathways were merged together in a parent pathway when the parent pathway sufficiently describes the merged child pathways. A list of all pathways can be found in Ref. 34: Supplementary data S3 for the upregulated and S5 for the downregulated proteins. A shortened list where all child pathways are excluded when a parent pathway is enriched can be found in Ref. 34: Supplementary S5 (upregulated) and S6 (downregulated), and the list for the Figure 2b and c in Ref. 34: Supplement S7 and S8, respectively.
As seen in Figure 2b, several pathways are enriched in the group of genes upregulated in senescence, such as translation, immune system, extracellular matrix, and metabolism. The numbers of pathways in the downregulated group were more modest and surprisingly included cellular senescence itself. In addition, the downregulated group included apoptosis regulating genes. It has previously been reported that apoptotic pathways are suppressed in senescence.
We next sought to compare the list of senescence related genes from U2OS cells reported here with those previously reported from other cell types. Alvelar et al. developed a comprenhensive database of genes associated with cellular senescence called CellAge. This integrative database utilizes a systems biology approach to the analysis of senescence and has developed gene expression signatures for cellular senescence.36 We compared our most significantly changed proteins to the CellAge senescence gene expression list. These results are shown in Figure 3 and in Table S9 in Ref. 34.
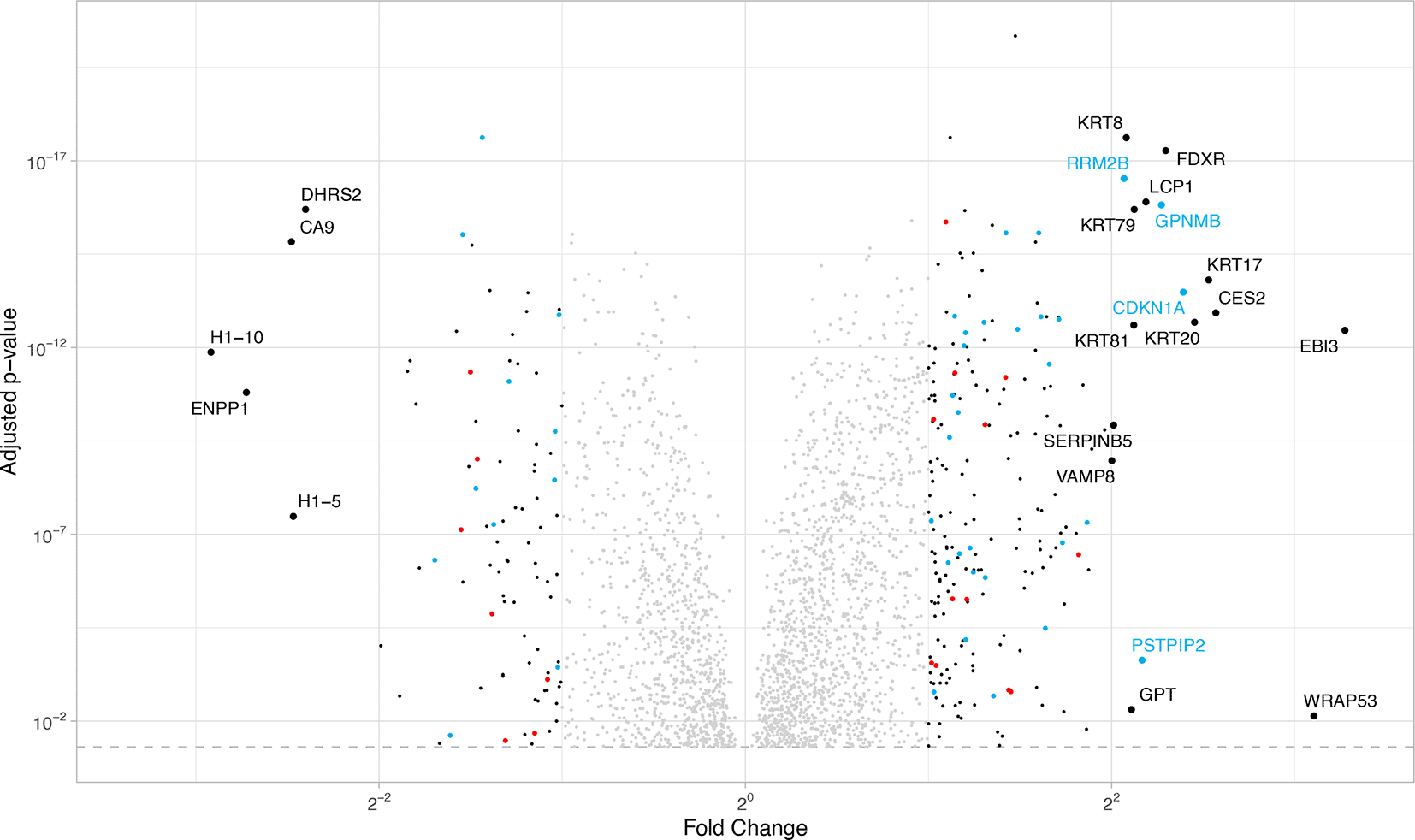
Proteins that have the same direction of regulation (over/under-expressed) in the CellAge database as well as in our data are marked in blue. Proteins with the opposite regulation (i.e. overexpressed in CellAge database but under-expressed in this study) are marked in red. A list with all significant CellAge/HAGR genes in our data can be found in Ref. 34: Supplementary S9.
Genes that have the same regulation pattern in our U2OS comparison as well as the CellAge list are marked in light blue, while genes that regulate differently (upregulated in one list and downregulated in the other one) are marked in red. Expressed genes (|log2FC| > 1) not included in the CellAge list are marked in black. For highly regulated genes (|log2FC|> 2) the gene name is added. As seen in Figure 3 the genes included in the Cell Age database, especially the highly regulated genes, act as expected. The genes that have a contrary regulation mostly have smaller fold changes. We also see several highly regulated genes in our data that are not included in the CellAge list, and are likely cell type specific senescence changes for U2OS cells.
The results from quantitative proteomics studies presented thus far were compared with those from RNA-Seq data generated under identical conditions using polyA selected mRNA. Following Illumina sequencing, quality control and further analyses including alignment, gene counts and differential expression, we retained only those genes with a mean count higher than 20 in the young and senescent conditions, fulfilled by 11835 genes. Figure 4 shows a comparison of the fold changes in RNA and protein measurements. In total, we had 4927 genes common between RNA and protein datasets. Grey dots depict genes that have a p-adjusted value above 0.05 and therefore do not have enough statistical power to be considered in the analysis. In this group there are 4122 genes. Genes with a p-value below 0.05 which change in both RNA and protein in the same direction (568 genes) are in black. Genes marked in red are statistically significant genes which are upregulated in senescence in RNA but downregulated in the protein measurements (74 genes). Genes marked in blue are regulated the opposite way, downregulated in senescence in RNA but upregulated in the protein measurements (163 genes). Genes with a in RNA or in protein of the antiregulated genes are labeled in Figure 4 a with gene name. A list with antiregulated genes in either direction can be found in Ref. 34: Supplementary Table S10. Genes with a in both conditions, RNA and protein, i.e. highly differentially regulated are few. In the red group, there is only one such gene, COMMD8, which is upregulated more than two-fold at the RNA level but downregulated more than two-fold in proteomics data. COMMD8 is a potential inhibitor of NF-KB signalling that controls senescence associated inflammatory signalling, hence this disparity in RNA versus proteomics data is interesting. In the blue group, there are nine genes that are highly upregulated at the protein level but more than two-fold downregulated in the RNASeq data. These are PHLPP1, ANLN, RACGAP1, KIF23, ITGB4, CCNB1, CAV1, WRAP53 and FAM83D with roles in insulin signalling, cell migration and cell growth.

(a) All genes are included that have a corresponding protein measurement. Grey means that the p-adjusted value was higher than 0.05 in RNA or protein. Black genes behave consistently in RNA and protein measurements. Red: A gene is up-regulated in the RNA but down-regulated in the protein measurements. Blue: Genes which are up-regulated in protein but down-regulated in RNA. Red and blue genes that have a in at least one condition are labeled. (b) Reactome pathways that are overexpressed in genes, which are up-regulated in proteins and down-regulated in RNASeq measurements. There are no enriched pathways for the genes that are down-regulated on protein and up-regulated on RNA level (red genes in a).
A Reactome analysis for the two gene groups with anticorrelated behaviour in Figure 4 was also carried out. No enriched functional pathways were found for the red group in Figure 4a). A summary of the Reactome results for the opposite condition, i.e. genes where RNA levels are downregulated but protein levels are upregulated (blue group in Figure 4a) can be seen in Figure 4b. The pathways and underlying genes can be seen in Table S14 in Ref. 34.
Cellular senescence is defined by irreversible growth arrest and profound changes in gene expression.2,37 Unsurprisingly, proteins involved in senescence and cell cycle progression are amongst the most highly up-regulated proteins in our data set. These include p21, a modulator of cell cycle progression and commonly employed marker of senescence, and RRM2B, a ribonucleotide reductase essential for DNA repair in non-proliferating cells38 (see Figure 2). In addition to the positive SA--Gal staining and identification of SAHF, the up-regulation of these proteins confirms the onset of cellular senescence in our model. Among the highly up-regulated pathways in the protein data (see Figure 2b), many are connected to SASP and cellular senescence. Furthermore, there are extracellular matrix alterations that are associated with cellular senescence.39 A further hallmark of cellular senescence is mitochondrial dysfunction which plays important roles not only in the senescence growth arrest but also in the development of SASP and resistance to cell death.40
Our data also shows that proteins commonly associated with cancer, including prognostic indicators such as SERPINB5, are significantly up-regulated in DNA damage-induced senescence. Examples include WRAP53, which is known to be over-expressed in a variety of cancer cell lines of different origins and promotes cellular transformation.41–44 The proteins KRT17,45 KRT8,46 KRT20,47 LCP1,48 and VAMP849 are also reported to be associated with cancer development/metastasis and cellular proliferation. Functional pathway analysis revealed terms related to translation, inflammation, mitochondrial dysfunction and cell migration to be significantly up-regulated; all of which are typical hallmarks of cancer cells in addition to some being common with senescence. Although considered a bona fide supressor of neoplastic transformation, our data suggests that senescence builds a transcriptional/translational landscape that may promote malignancy. Interestingly, we observe a significant enrichment in proteins involved in rRNA processing which is in contrast to previous studies.50 Ribosome biogenesis and protein translation are finely coordinated and essential for cell growth, proliferation and differentiation. Multiple RP proteins have extra-ribosomal functions including activation of pathways in response to stress, resulting in cell cycle arrest and apoptosis. In cancers, these functions are often misregulated.51 Several studies have provided evidence for active keratin involvement in cancer cell invasion and metastasis. The keratinization pathway is here driven by the highly up-regulated KRT proteins.52 Tissue remodeling is promoted by MET tyrosine kinase receptor, which underlies developmental morphogenesis, wound repair, organ homeostasis and cancer metastasis.53 Several other upregulated pathways are related to cancer. Among these is the metabolism of amino acids and derivatives (GPT), which is here driven by the RPL and RPS gene group and the GTP group of our highly expressed genes.
The most significantly down-regulated proteins in our dataset include DHRS2, H1, and ENPP1. Decreased expression of DHRS2 contributes to p53 stabilization thus promoting the onset of senescence. In mice, Enpp1 has been shown to play a crucial role in regulating aging via Klotho expression and its down-regulation has been shown to be associated with aging.54 We were initially surprised to find the cellular senescence term to be significantly down-regulated in our data set (see Figure 2c). Among the genes in this pathway is MAP 2K6, which is involved in stress-induced cell cycle arrest, transcription activation and apoptosis.55 Another significant example is ERF, which is involved in development, apoptosis, and regulation of telomerase, a key regulator in age-related or replicative senescence. We believe this change may reflect a very late stage of senescence wherein the expression of pro-senescence proteins has reduced and which may represent an incomplete senescent phenotype.
To benchmark this study, we compared the proteomic results with known datasets on senescence through the Cell- Age database.36 Generally, our data is in agreement with that published by Alvelar et al. We believe that any discrepancies observed are likely to be due to differences in cell line and method of senescence-induction, suggesting that this is a useful resource that can support other studies on senescence in this model.
A large number of pathway terms relating to translation and rRNA processing appeared amongst the proteins up-regulated in senescence in U2OS cells. This led us to compare proteomic data with RNA-Seq which highlights significant irregularities between the detected level of proteins and the expression of their corresponding genes suggesting altered mechanisms of translational regulation between proliferating and senescent cells. Interestingly, our data highlights that genes/proteins where RNA and protein levels are highly anti-regulated (Figure 4a) are typically associated with aging and senescence. Examples include PHLPP1, which protects against age-related intervertebral disc degeneration,56 and ANLN, the depletion of which induces cellular senescence.57 Expression of ITGB4 is reportedly down-regulated under oxidative stress or upon inflammatory stimulation leading to the induction of senescence in epithelial cells, mediated through p53 activity.58 CCNB1 silencing inhibits cell proliferation and promotes cell senescence via activation of the p53 signaling pathway in pancreatic cancer,59 CAV1, which has been shown to induce senescence in resting human diploid fibroblasts,60 and WRAP53 and FAM83D, which are commonly over-expressed in a variety of cancers and known to trigger apoptosis.41
The pathways driven by genes which are up-regulated at the protein level but down-regulated at the RNA level are varied and involve multiple cellular processes. An interesting pathway comprises genes associated with the assembly of the primary cilium, a sensory structure that interprets extracellular signals to stimulate a range of cellular pathways including growth, response to nutrient deprivation, and cellular development.61
Additional examples include proteins involved in a range of signal cascades and the stimulation of transmembrane receptors. Depending on the cellular context, this may impact cellular proliferation, differentiation and survival. Additional pathways following the same pattern including those related to autophagy, DNA repair and those related to cellular senescence. Cytokines (key components of SASP) and inflammatory mediators are an interesting example of proteins that play a significant role in the senescent phenotype but exhibit striking differences between mRNA and protein levels. These observations may suggest that mechanisms of translational regulation play a more significant role in the induction of the senescent phenotype than transcriptional regulation alone.
We have employed a cellular model of senescence in a cancer derived cell line to investigate broad transcriptomic and proteomic changes. By combining RNA-seq and proteomic datasets, our data demonstrate a dramatically altered translational landscape at both the protein and RNA level, whilst demonstrating that the model exhibits all the characteristics and markers of the senescent phenotype. The model can be easily implemented and utilised to study senescence in cancer cells in a wide range of contexts. Our data reveal a range of age and disease-relevant proteins and pathways that are altered in senescent cancer cells, such as the assembly of the primary cilium, and highlights the emerging role of lipids in the senescent phenotype. Our data are also suggestive of very significant regulatory changes in translation that warrant further investigation.
ENA.EMBL: RNA-Seq data for young and senescent U2OS cells. Accession number RJEB59999. https://identifiers.org/ena.embl: PRJEB59999
Conceptualization by TS, MA-R, SNG. Data Curation was done by TS and FG. Funding Acquisition, Supervision and Project Administration was undertaken by SNG. Investigation was done by TS and MA-R. Methodology was developed by FG and SNG. Computational analysis was carried out by FG. Visualization was done by MA-R and FG. The original draft was written by by MA-R and FG and subsequent review editing by TS and SNG.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)