Keywords
Crassostrea virginica, Microbiome, Metagenomics, Bead-beating
This article is included in the Bioinformatics gateway.
Crassostrea virginica, Microbiome, Metagenomics, Bead-beating
Crassostrea virginica, commonly called as the Eastern oyster or the American oyster, are represented in as much as 75% of the commercially harvested oysters in the United States (US), especially along the coastal regions of Florida, Texas, Louisiana, Mississippi, and Alabama; these states contribute to more than 60% of the total oyster harvest in the US.1 Oysters are valued not only as seafood, but also for their myriad of services rendered to their native ecosystems, such as enhancing water quality,2 bioextraction of dissolved nitrogen and other pollutants3 as well as providing refuge and foraging habitat for marine organisms, such as smaller mollusks, invertebrates, fish and crustaceans,4 within the oyster reefs.5 Due to their filter feeding mechanism, oysters have been shown to concentrate estuarine microbiomes within their gills and digestive tracts and some microbes remain symbiotically associated with the oyster host species, called as the autochthonous microbiota.6–8 Interestingly, the autochthonous oyster-associated bacteria are now beginning to be better understood for their beneficial functions, some of which include assisting with digestion processes,9 providing the bivalve host with a suite of growth-promoting vitamins and amino acids,10 protection of hosts from pathogens by producing antimicrobials, or by forming an outer biomass barrier thus discouraging colonization by other strains11 and even facilitating shell formation by the extrapallial fluid microbiome communities.12
Proteobacteria, Cyanobacteria, Bacteroidia, Mollicutes, Bacteroidetes, Tenericutes and Firmicutes are some of the major phyla that have been documented from the Eastern oysters.12–18 Among the genera that are commonly found associated with the Eastern oysters are pathogens such as the Vibrio species; nonpathogenic bacteria that are beneficial to the oysters have also been identified including Mycoplasma, Pseudoalteromonas, Burkholderia, Bacteroides, Lactobacillis, Acetobacter, Allobaculum, Ruminococcus, Nocardia, and Oceanospirillales.6,19 Our previous studies found Cyanobacteria accounting for 50-75% of the total microbial communities, based on 16S rRNA amplicon metagenomics.20 In another microcosm-based study related to the Deepwater Horizon oil spill, Pseudomonas spp. were the dominant bacteria in the Eastern oysters.20–23 More recently, our metagenomics surveys of the Eastern oysters indicated the dominance of Psychrobacter spp., along with other members that included Lactobacillus, Burkholderia, Bradyrhizobium, Afipia and Delfitia.
However, there are factors that can confound the microbial community analysis, especially interference by the host tissues and cellular by-products, sample type and preparation,24 along with other factors such as different DNA extraction methods25–27 and even the bioinformatics pipeline used for the taxonomic analysis.28 For example, Zhang et al.29 compared four different DNA isolation methods on commercial oysters, that included two commercial kits and two traditional lysis methods: the phenol-chloroform and the boiling methods. While the commercial kits performed better in terms of the DNA quality, the phenol-chloroform protocol had a high DNA yield; besides, the boiling method, though simple and cheap, required some sample enrichment to detect the food-borne pathogens. Another related study reported that the features of tissue type, sampling, and method of purifying nucleic acid had impact of the Pacific oyster’s microbial compositions.30 Note that most of the commercial DNA extraction kits rely on a combination of chemical and physical lysis (bead beating) to release nucleic acids from complex environmental samples and the oyster samples are further exacerbated by the presence of sticky mucopolysaccharides and other sticky proteins and biochemical compounds that impede both quality and quantity of DNA isolation. We have typically used the DNeasy PowerLyzer PowerSoil DNA extraction kit paired with the FastPrep sample disruption instrument designed to quickly and efficiently homogenize, grind and lyse biological samples, to study the oyster-associated microbiomes. This DNA extraction protocol entails adding samples to individual lysing matrix tubes provided in the DNA isolation kit and placed into sample holder adapters within the instrument. The unique, optimized motion of the FastPrep-24 5G shakes the samples in a tridimensional motion to result in the collision of the cells in the sample matrix with the lysing particles. Additionally, the Bead Ruptor Elite from Omni International has been demonstrated to perform well for host microbiome studies. The Ruptor Elite is a bead mill homogenizer and is specifically designed for grinding, lysing, and homogenizing biological samples prior to molecular extraction. Therefore, the main goal of this study was to compare the two different lysis protocols: 1) FastPrep versus 2) the Bead Ruptor Elite method and evaluate the bacterial communities in oysters collected over three separate time points. Even though both protocols rely on the same method of bead mill-based homogenization and lysis of biological samples, clear differences emerged in the taxa identified from the FastPrep method relative to the Ruptor Elite method. This suggests that several DNA extraction protocols and lysis methods should be evaluated for the assessment of bacterial communities in their entirety, especially when complex environmental samples are involved, such as the oyster tissues.
Oysters were collected from the aquaculture research lease of the Wakulla Environmental Institute located in Oyster Bay [30.0342702, -84.3558844], near Panacea, Florida. Oysters were grown in plastic cages, suspended from lines attached to pilings at depths of 1-2 meters (Australian Adjustable Long-Line System). At each sampling period (December 2020, January 2021 and March 2021), n=15 adult oysters of approximately the same size were collected over ice and transported to the laboratory at FAMU, where they were rinsed, shucked, tissues harvested and stored at 4°C for the DNA extraction to be carried out the next day. Genomic DNA from minced oyster tissues were extracted using DNeasy PowerLyzer PowerSoil DNA extraction kit was according to the manufacturer’s protocol but the samples were processed in two separate batches: one that was processed for lysis using the MSP FastPrep method (samples labeled from 1-6) and the second set was processed using the Bead Ruptor Elite method by Omni International (samples labeled as A-F), respectively. In this manner, six DNA extracts were obtained for each group (Table 1), and the extracted DNA was analyzed for quality and quantity using a micro-volume spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
We used the 515F/926R primer pairs, containing the CS1 and CS2 common sequence linkers (Walters et al.31), to amplify the V4-V5 variable regions of the 16S rRNA gene. The PCR amplicon generation was performed in two independent steps: ST1 and ST2, as described previously.32 The first step (ST1) basically involves the generation of the amplicon products and the attachment of the common sequence linkers (CS1 and CS2) at the 5’ end of all the DNA molecules. The reaction mixture in this step comprised 9 μL of the PCR reagent and 1 μL of the Genomic DNA template. The reagent comprised 5 μL repliQa HiFi ToughMix (PCR mastermix, Quantabio), 3μL DNA-free water, and 0.5 μL each of the gene specific forward and reverse primers containing the 5’ linkers. The ST1 PCR condition comprises initial denaturation at 98°C for 2 min, followed by 28 cycles of denaturation at 98°C for 10min, annealing at 50°C for 1 min, and elongation at 68°C for 1 min.
The second step PCR (ST2) was basically to incorporate sequence adapters and sample-specific barcodes into the ST1 amplicons. The ST2 reaction mixture comprised 3 μL of repliQa HiFi ToughMix (PCR mastermix, Quantabio), 4 μL of DNA-free water, 1 μL each of the forward and reverse sample-unique Access Array Primer pairs (Fluidigm, South San Francisco, CA; Item# 100-4876), and 1μL of ST1 PCR product, as the template. The forward Access Array primer contained the 3’ common sequence linker (CS1), and the adapter sequence, while the reverse Access Array primer contained the sample-specific barcode in addition to the 3’ CS2 and the adapter sequence. The ST2 PCR condition was like the ST1, only that the ST2 had 8 cycles; besides, the annealing process of the ST2 was performed at 60°C.
The amplicons were pooled, and the library preparations and sequencing using the Illumina MiSeq based on V3 chemistry (600 cycles) were carried out at the Genomic and Microbiome Core Facility at Rush University. Meanwhile, to address the issue of low sequence complexity with 16SrRNA amplicons, the pooled library was spiked illumina Phix viral DNA library (a prepared high complexity library) at a 15% volume ratio.
The MicrobiomeAnalyst pipeline was applied for the relative abundance analysis and to create the metagenomics plots using data processed from QIIME2.
The metagenomic sequences obtained from the FastPrep and the Bead Ruptor Elite samples have been deposited in NCBI’s Sequence Read Archive under BioProject PRJNA986007 with the accession numbers SRR25013903 and SRR25013902, respectively, and BioSamples SAMN35839752 and 35839753.
The extracted DNA was quantified using a micro-volume spectrophotometer (NanoDrop Technologies) and the results are show in Table 1. The statistical significance of difference in DNA concentrations between DNA isolated using the FastPrep lysis (MP Inc.) versus the Bead Ruptor Elite (Omni international Inc.) methods was assessed with the Mann-Whitney U test.33 The distribution of the DNA quantities was not normal, and the value of the variable was continuous, making a non-parametric test suitable for the analysis. The Mann-Whitney U test is used to determine whether there is a statistically significant difference between the medians of two independent groups. The Mann-Whitney U statistic was selected as the smallest of the two U values for the two groups calculated following the U statistic formula,33 and the U value was compared with the standard Mann-Whitney U critical value. At 0.05 significant level, the U statistic was 11 while the U critical was 5, suggesting there was no evidence to support the null hypothesis, which states that the DNA concentrations from the two groups were equal (Table 2). In other words, the quantity of DNA was affected by the extraction protocol. This finding mirrors previous reports, which suggests that the nature of DNA lysis equipment can be a significant factor with impacting the outcome of metagenomics studies. For example, Zhang et al.34 reported that the bead-beating intensity in DNA lysis affected the DNA yield in gut samples. In line with this report, our results also suggest that the FastPrep lysis method provided the desired beating intensity than the Bead Ruptor Elite, hence the average DNA yield from the FastPrep was higher than the Bead Ruptor Elite.
The DNA isolation kit used was the same with the only variation being the lysis method.
“L” stands for the “Letters” (MP FastPrep) and “N” stands for the “Numbers” (Bead Ruptor Elite) group.
| Values (ng/mL) | 2 | 2.2 | 2.6 | 3.01 | 3.9 | 4.6 | 6.2 | 7.1 | 7.2 | 9.9 | 16.2 | 31.8 |
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Group | L | L | L | N | L | N | N | N | N | L | L | N |
| RankL | 32 | |||||||||||
| RankN | 46 | |||||||||||
| U statistic | 11 | |||||||||||
| U Critical (α=0.05) | 5 |
Metagenomic-based microbial taxonomic analysis of the eastern oyster
The actual abundance of microbiomes in the oyster tissue at the phylum level is shown in Figure 1. Vertical bars represent the abundance of organisms in each sample and the colors represent the identified phyla. The samples were mostly populated by the Protobacteria phyla, regardless of lysis method. The samples treated by the FastPrep lysis method also harbored Firmicutes and Bacteroidota, which were not identified in the Bead Ruptor Elite lysis method. Traces of Actinobacteria were found in both groups. This is indicative that the DNA extraction was impacted by the lysis method such that MP FastPrep is a better choice for the oyster tissue samples used in this study. The absence of the Gram-positive bacteria, Firmicutes, from the Bead Ruptor Elite group indicates that this method could not provide the adequate bead beating intensity required to break the complex cell wall of Gram-positive bacteria.34 On the other hand, abundance of Bacteroidetes and Firmicutes in the FastPrep group strongly indicates that this method could lyse both the Gram-negative and the Gram-positive bacteria in oyster samples.34
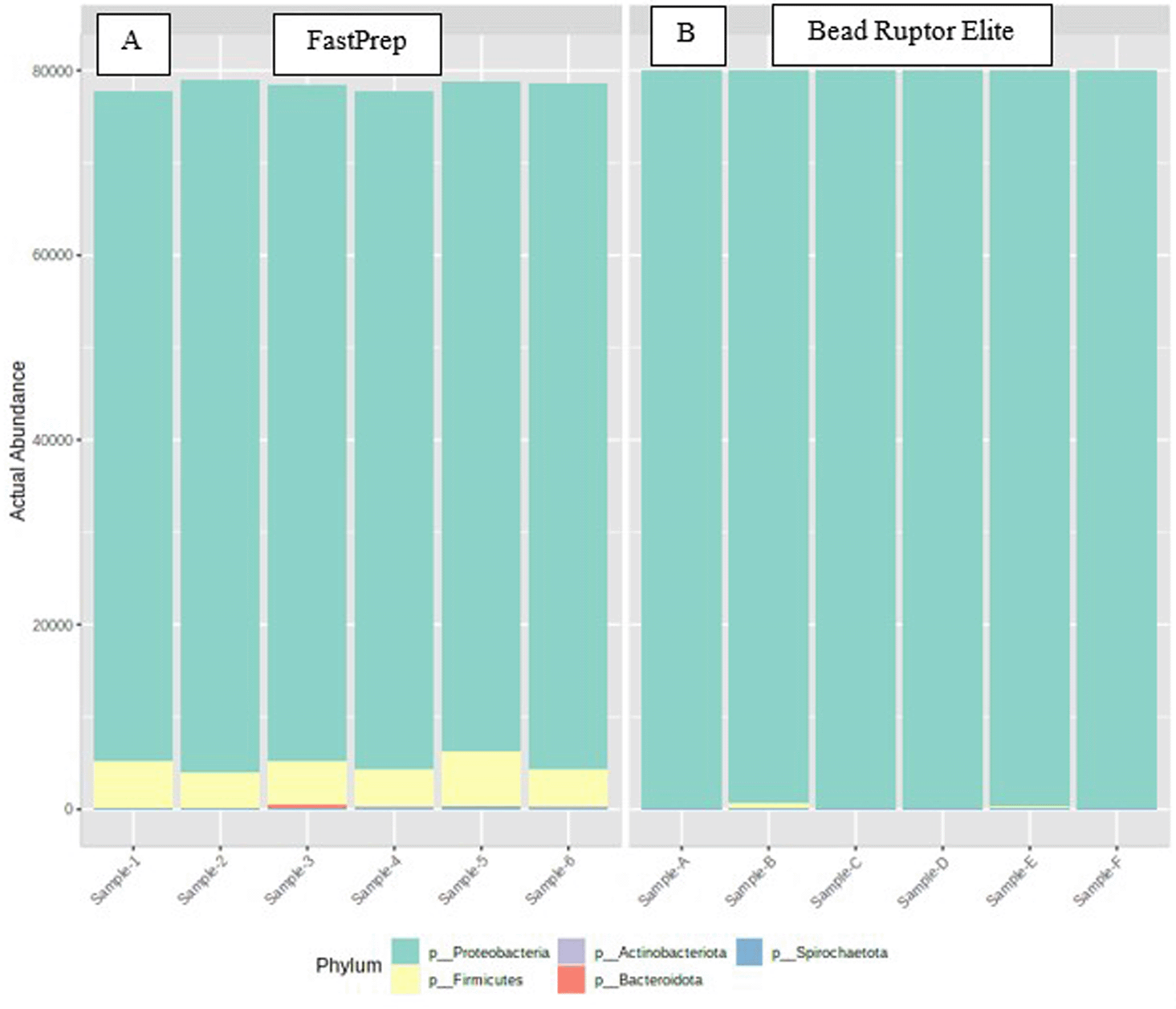
Samples were isolated by the same DNA extraction protocol but differed in their lysis method: A, when samples were lysed by the FastPrep method and B, when samples were lysed by the Bead Ruptor Elite method.
The genus-level taxonomic groups identified are shown in Figure 2, which showed that the Delftia genus dominated up to 80-85% of all the genera identified across all samples when the lysis was performed using the FastPrep method. Interestingly, when the lysis was carried out using the Bead Ruptor Elite lysis method, the predominant genera at 80-90% was Burkholderia_Caballeronia_Paraburkholderia. Mycoplasma was another genus identified from the samples treated with the FastPrep method but were mostly absent in the Bead Ruptor Elite treated samples. This shows that two different lysis protocols can result in different taxonomic data and thus researchers need to be cognizant of DNA extraction process being followed for such metagenomics studies, especially those that involve host tissues containing a high amount of mucous and other inhibitory materials. Both Delftia and Burkholderia belong to the Betaproteobacteria class and Pseudomonadota phylum. However, the bacterial genera identified from the FastPrep method were more diverse, thus suggesting that this lysis method provided a better recovery of the oyster microbiota than the Bead Ruptor Elite method. In addition, the presence of Burkholderia, a Gram-negative bacteria with a fairly easy-to-lyse cell wall, in the Bead Ruptor Elite group further suggests that this method could not provide enough intensity to rupture the cell walls of more robust bacteria in the oyster samples, lending further support that DNA lysis can significantly impact the outcome of metagenomics assessment, especially in complex samples from host tissues and the gut ecosystem.34
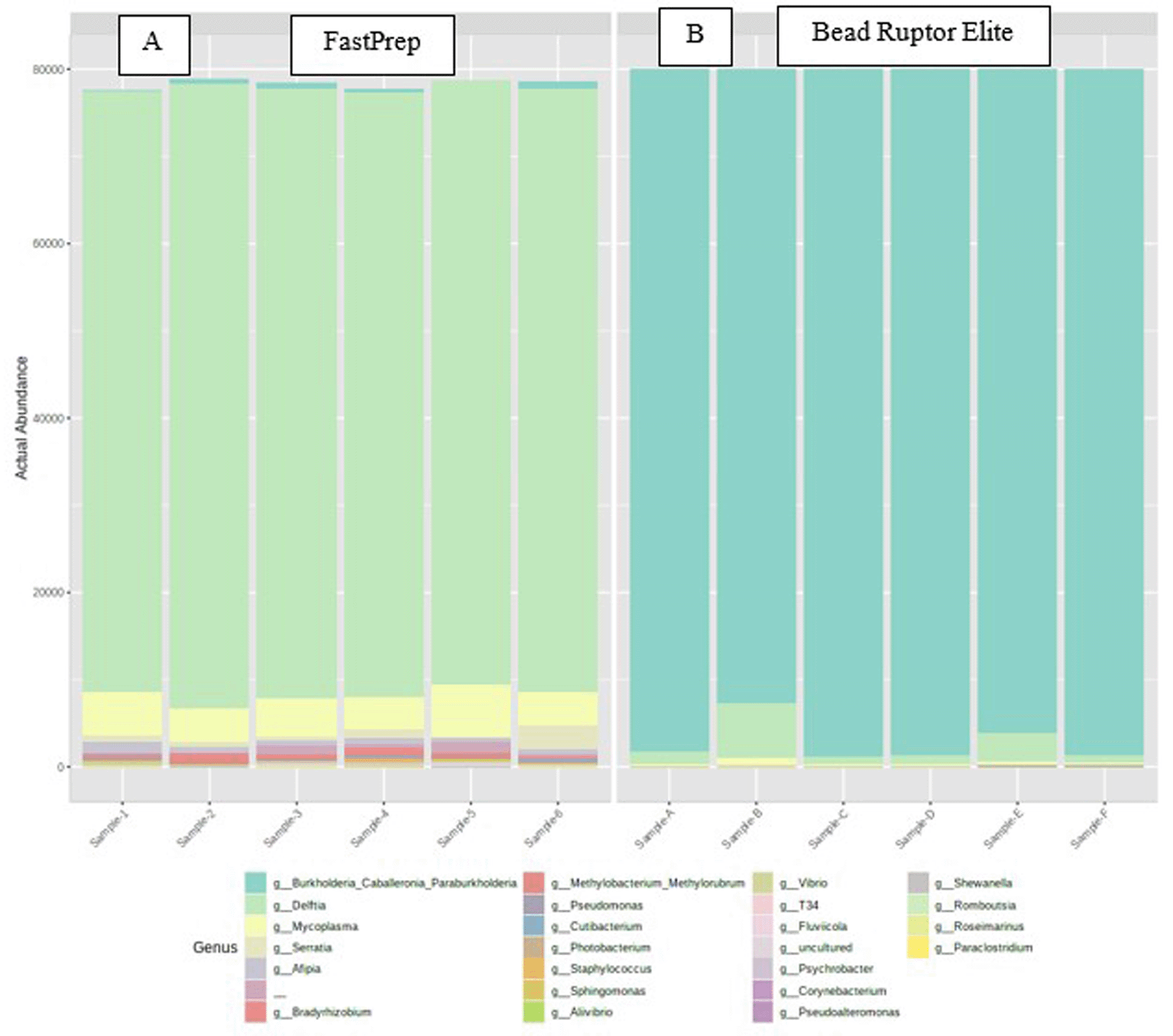
Samples were isolated by the same DNA extraction protocol but differed in their lysis method: A, when samples were lysed by the FastPrep method and B, when samples were lysed by the Bead Ruptor Elite method.
Delftia is a genus of Gram-negative bacteria named after the city of Delft in The Netherlands, where the genus was first isolated. Delftia is represented by five recognized species, including D. acidovorans, D. tsuruhatensis, D. lacustris, D. litopenaei, and D. deserti. Delftia species have wide geographical distribution and they have been found in fresh and marine water, plants, soil, and sludge.35 Banach et al.36 found Delftia to be associated with aquatic ferns and synthesized plant growth-promoting substances. Delftias are non-sporulating and are known for their unique metabolic ability to break down or transform different forms of environmental pollutants. For example, De Gusseme et al.37 reported that Delftia tsuruhatensis and Pseudomonas aeruginosa were solely responsible for the total removal of acetominophen, an analgesic and antipyretic drug, from a wastewater treatment plant. According to Zhang et al.,29 Delftia tsuruhatensis can degrade several chloroanilines through the process of mineralization, which is an important step in nitrogen cycle. Wu et al.38 reported that a strain identified as Delftia lacustris was resistant to heavy metals, such as Cr, Pb, Hg, and Cd. They also reported that the strain could degrade aromatic compounds like naphenol, naphthalene, 2-methylnaphthalene, and toluene. Aside from the ability to remove pollutants, Delftia species can play significant roles in nitrogen cycling processes: Delfta tsuruhatensis performed both heterotrophic nitrification and aerobic denitrification functions in a sewage treatment ecosystem.39 Chen et al.40 isolated a heterotrophic and aerobic nitrifying-denitrifying strain of Delftia tsuruhetensis from a co-culture of rice husk and swine manure. The strain had ammonium removal rate of about 98% from a piggery wastewater. We are tempted to predict that Delftia species likely play several roles related to ecosystem services withing the Eastern oysters.
The genera Burkholderia is a Gram-negative bacterium of the Betaproteobacteria class with about 80 recognized species. Burkholderia has diverse bioremediation properties and can synthesize a wide range of antimicrobial compounds.41 Interestingly, Burkholderia predominated amongst the marine sponge-associated bacteria.42 It has also been shown that Burkholderia species are active in the denitrification processes. Vitale et al.43 stated that sites occupied by Burkholderia are characterized by limited oxygen where the organisms generate energy by denitrification; furthermore, several genes required for growth under denitrification condition abound in Burkholderia thailandensis.
Two types of diversity analysis were applied to estimate the species distribution within the community based on operational taxonomy units (OTUs). The observed diversity or richness is a measure of the individual number of species found in each sample. The richness does not account for the relative proportion or abundance of the individual species in a sample. In Figure 3(A), the OTUs from the FastPrep method were about 40% more diverse than the OTUs from the Bead Ruptor Elite method. The observed difference in OTUs suggests that the DNA extraction kits significantly impacted the richness of the oyster tissue microbiome community.
Similarly, the Shannon diversity or equitability index considers the proportion or evenness of species in a community. The Shannon index ranges from 0 to 144; a perfectly distributed community takes the index of 1. The Shannon equitability of the OTUs from the two DNA lysis methods kit is shown in Figure 3(B). By this measure, species from the FastPrep method were about 70% more evenly distributed than the Bead Ruptor Elite method. In other words, the FastPrep method resulted in richer and evenly diversified bacterial community relative to the Bead Ruptor Elite method.
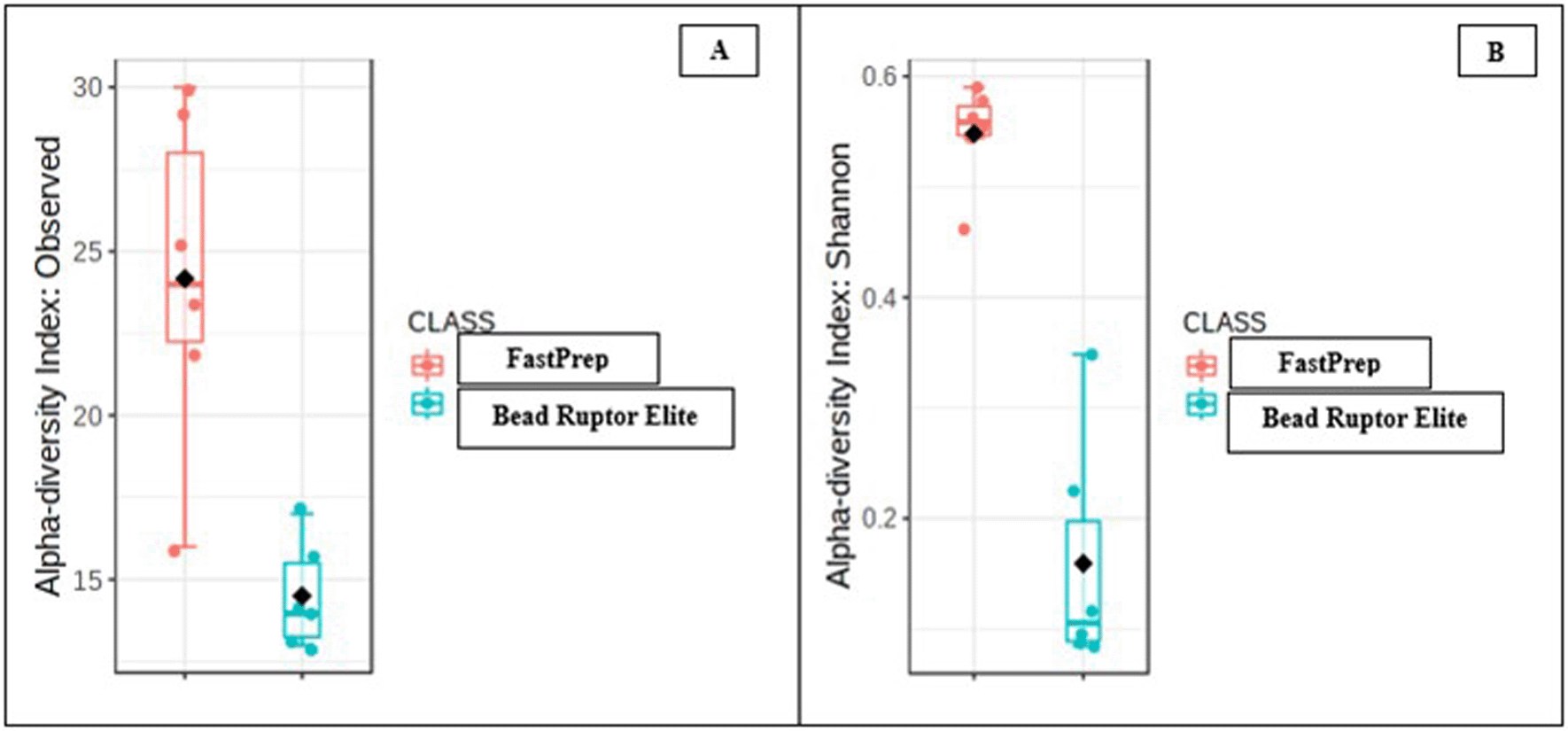
Shown are A, the observed diversity index and B, the Shannon equitability index.
The plot of the principal coordinate analysis of the species from the oyster tissue samples grouped by the two DNA lysis extraction protocols is shown in Figure 4. The first principal coordinate axis accounted for 54.8% of the total variation, while the second principal coordinate axis accounted for 16% of the total variation. Projecting the sample points on the first principal coordinate axis (the horizontal axis), the bacterial communities from the FastPrep method were completely separated from the Bead Ruptor Elite method, indicating that the DNA extraction method significantly affected the identification of bacterial species from the oyster tissue samples.
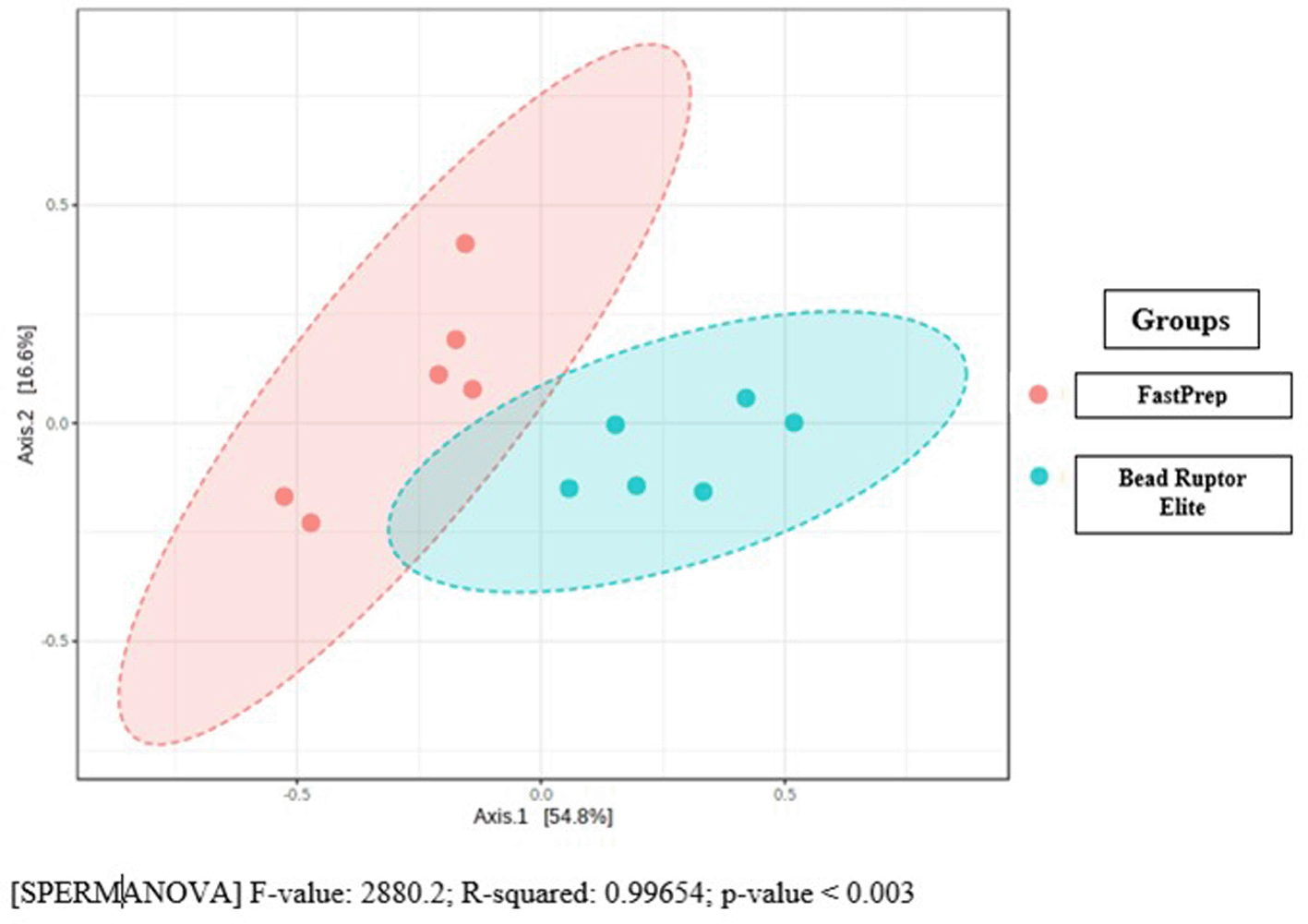
[SPERMANOVA] F-value: 2880.2; R-squared: 0.99654; p-value < 0.003.
The accompanying permutational multivariant analysis of variance (PERMANOVA) was carried out to investigate the statistical significance of the difference in microbial compositions from the two groups of oyster samples as a function of the DNA lysis protocol, such that the predictor of the species diversity model was the DNA extraction method. The result indicate that the R2 value was 0.9965, p value of < 0.003, meaning that the effect of DNA extraction method was significant and that it accounted for about 99.7% of the total species diversity in the two groups.
A heatmap showing the variability of identified microbiome at the family level across different oyster tissue samples is shown in Figure 5. Specifically, this heatmap analysis indicated Burkholderia species to be overrepresented in the Bead Ruptor lysis method, which goes well with the findings that these were the predominant genera at 80-90% abundance using this lysis method. Similarly, Pseudoalteromonadaceae were most abundant in the Bead Ruptor lysis method. Overall, Figure 5 shows those bacteria in red arrows that were overrepresented in the Bead Ruptor Elite method and blue arrows represent bacterial communities that were found to be overrepresented in the FastPrep method, respectively.
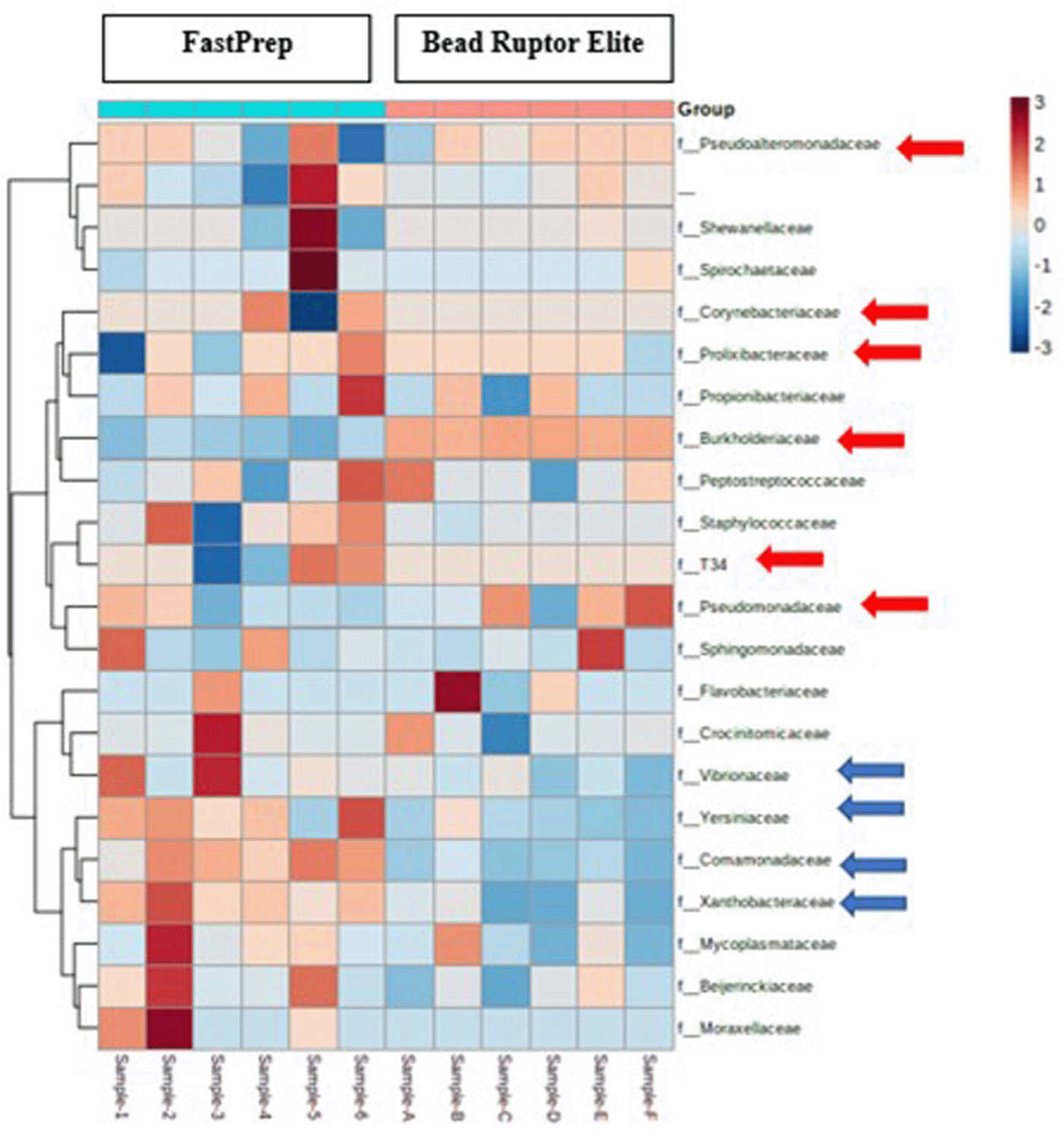
Each column represents a sample, and each row represents a bacterial family. Red arrows represent the overrepresented bacteria in the Bead Ruptor Elite method and blue arrows represent bacterial communities that were found to be overrepresented in the FastPrep method.
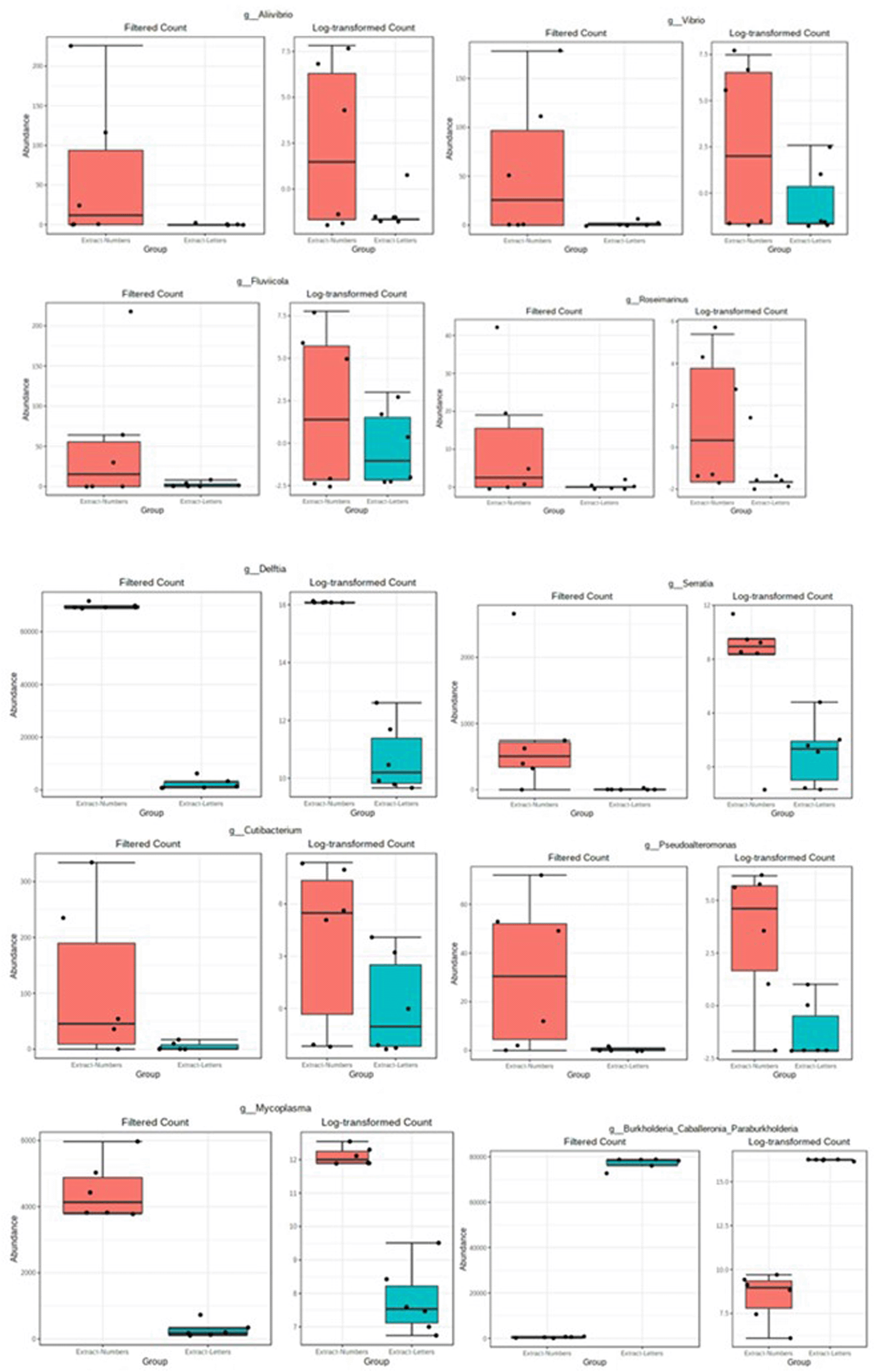
The result of the MetagenomeSeq differential analysis is shown in Figure 6. Ten different genera from the entire samples were differentially abundant between the two lysis methods tested. These included Alivibrio, Vibrio, Fluvicoda, Rosemainus, Delftia, Serratia, Cutibacterium, Pseudoalteromonas, and Mycoplasma genera were differentially abundant in the FastPrep method. Conversely, Burkholderia-Cabelleronia-Paraburkholderia bacterial group was differentially abundant in the Bead Ruptor Elite method. In other words, the DNA extraction lysis protocol significantly influenced the type and abundance of microbial genera in the oyster tissue samples.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)