Keywords
Methicillin Resistant Staphylococcus aureus (MRSA), Penicillin binding protein 2A (PBP2A), Diagnostic kit, Antigen, Multi-epitope
This article is included in the Cell & Molecular Biology gateway.
Methicillin Resistant Staphylococcus aureus (MRSA), Penicillin binding protein 2A (PBP2A), Diagnostic kit, Antigen, Multi-epitope
Methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infection (BSI) remains a major cause of concern in health systems all over the world, because of the high incidence rates, which correlate positively with significant upward trends in patient age and comorbidities, longer length of stay, access to health care, both non-intravascular and vascular catheter sources, and the associated high morbidity and mortality rates in infected patients (Gasch et al., 2011, 2013). MRSA BSI also occurs in the community, more commonly in patients with a previous MRSA colonization, injection drug use, or who are admitted from a skilled nursing facility, who have had surgery in the previous year, or are on dialysis during hospitalization, or have bone/joint as a common source of CA-BSI (McLaughlin and Smith, 2019; Gouin, 2019).
Furthermore, the hematogenous spread of MRSA in 10-20% of BSI patients is of major concern, causing secondary complications such as abscesses, vertebral osteomyelitis, endocarditis and implant associated infections of prosthetic joints, electronic cardiac devices, and so on, which can occur weeks or months after the primary infection (Tande et al., 2016).
Methicillin resistance in Staphylococcus aureus is mediated by PBP-2a, a penicillin-binding protein produced by the mecA gene that enables the organism to proliferate and replicate when beta-lactam drugs like methicillin are present. The mecA gene is located on a mobile genetic element called staphylococcal chromosome cassette (SCCmec) (Archer et al., 1994). Though there are antibiotics of choice for treating bloodstream infections due to MRSA; vancomycin and daptomycin (Liu et al., 2011), treatment failure has been associated with lower (<15 mg/l) trough levels of vancomycin (Forstner et al., 2013), reduced vancomycin susceptibility (Howden et al., 2004) and growing incidence of “hetero-resistant” vancomycin intermediate susceptible S. aureus (hVISA) strains (Hiramatsu et al., 1997). Thus, the occurrence of MRSA isolates with decreased susceptibility to vancomycin has grown over the past few years, most likely also as a result of patients with MRSA infections using vancomycin more frequently (Hiramatsu, 2001).
Except for patients with pneumonia, daptomycin is still a viable therapy choice for these MRSA BSI isolates (Moise et al., 2009). As a result, insufficient source control, an uncertain primary focus of infection, or insufficient antibiotic therapy are additional risk factors for BSI consequences (Seifert et al., 2008).
The impact of MRSA infection prevention on patient outcomes and length of hospital stays has been significantly reduced by adopting tactics including monitoring for MRSA carriers upon arrival, followed by isolation and/or decolonization, and hand hygiene (Kavanagh, 2019).
However, up to the present moment, MRSA vaccination attempts have proved unsatisfactory. Phase IIb/III trials have been advanced for three candidates. Bivalent conjugate vaccine StaphVAX, which targets capsular polysaccharide types 5 and 8, failed to produce long-lasting protection (Shinefield et al., 2002). The experiment was prematurely terminated because V710, a monovalent vaccination that targets the iron salvage protein IsdB, was actually linked to higher mortality (Giersing et al., 2016). Most recently, Pfizer reported that the phase IIb trial of their multi-antigen vaccine (PF-06290510) has been suspended due to futility in December 2018 (Pfizer, 2019).
Patients with MRSA infection may require a longer and more expensive course of therapy and recuperation, if their infection is not adequately identified in the early stages of illness progression (Cosgrove, 2006). Any infection control monitoring approach that aims to reduce the exposure of healthcare personnel, other members of the community, or other patients to this disease must include an accurate and prompt diagnosis of the MRSA status. The conventional microbiological culture techniques including the detection of resistance to methicillin among staphylococcal isolates, based on phenotypic assays such as a disk diffusion test, minimum inhibitory concentration determination or broth microdilution testing are assumed to be 100% reliable but require quite some time (up to 5 days for set up, data collection and analysis (Palavecino, 2020). Similarly, there are commercially available chromogenic agars that can be used for MRSA identification with read out in between 24 and 48 hours (Lee et al., 2013). The real-time PCR assay is regarded as the global gold standard for diagnosing MRSA, increasingly the tool of choice to help clinicians understand MRSA and S. aureus infections and to guide patient care; generally faster than the traditional culture methods (by saving 48-72 hours). However, while most real-time PCR methods offer similar sensitivity, they present with different specificity leading to reports of unacceptably high number of MRSA false negative results—ranging from 6% to 30%—and false positive rates as high as 20% (Jeyaratnam et al., 2008; Rossney et al., 2008).
It is basically understood that MRSA and S. aureus misdiagnoses can result in poor patient management and care delivery as well as serious primary and secondary consequences, including fatalities (Nathwani et al., 2008; Diekema and Saubolle, 2011; Olatunde et al., 2022; Owolabi and Olatunde, 2022; Olatunde and Owolabi, 2022). Therefore, the severity of the damage these strains can bring to infected patients and the potential for community dissemination, infections caused by multidrug-resistant bacteria like MRSA necessitate rapid and accurate diagnosis and elimination at an early stage (Rahman et al., 2018). Immunopeptidomics is a field of study that examines the collections of all peptides connected to human leukocyte antigen (HLA) molecules, and it is frequently fueled by mass spectrometry (MS) technologies. In recent years, several in-silico approaches have been used to predict and design multi-epitope peptides capable of bounding to major histocompatibility complexes (MHC) (Paul et al., 2021; Moradi et al., 2022; Oladipo et al., 2022; Khan et al., 2022; Ghulam et al., 2022; Islam et al., 2022). This study sought to develop a B-cell multi-epitope peptide for the diagnosis of MRSA infection from the blood of individuals with MRSA blood stream infection using an immunoinformatics approach, which may aid in immune response and improve serodiagnosis accuracy while speeding up diagnosis, particularly for those residing in developing and endemic areas. The data and findings from this study could be used as starting points for the designing of improved infection diagnostic methods.
Figure 1 shows the process flow for the steps performed throughout this investigation.

This figure is an original figure produced by the authors for this article.
We identified some highly conserved and experimentally verified potential antigenic penicillin-binding protein 2A (PBP2A) from the genomes of Methicillin Resistant Staphylococcus aureus (MRSA), consisting of ALJ10988.1, ORN56903.1, AFJ06714.1, AEO00772.1, WP_000721309.1, WP_057521704.1, and WP_063851348.1. The National Center for Biotechnology Information (NCBI, Retrieved February 23, 2023) was utilized to retrieve the protein sequences. After running a query with their accession numbers, the protein sequences were obtained as a FASTA format.
Using the Vaxijen V2.0 server program (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova and Flower, 2007b), the antigenicity of the retrieved protein sequences was evaluated at a threshold value of 0.5. The Vaxijen investigates the antigenic protein’s capacity to bind to or interact with B-cell receptors.
TMHMM Server 5.0 (https://services.healthtech.dtu.dk/services/TMHMM-2.0/) was used to predict the transmembrane structure of proteins. In order to evaluate if the proteins are constituted within the membrane or not, the protein sequences were uploaded into the server window and three regions (S), comprising the inner and outside transmembrane were examined (Möller et al., 2001; Khazaei and Moghadamizad, 2022; Shams et al., 2021). Additionally, SignalP Server (https://services.healthtech.dtu.dk/services/SignalP-5.0/) investigated the signal peptide prediction of the membrane proteins to ascertain whether the proteins found within the membrane were plasma membrane or subcellular (Almagro et al., 2019).
Setting all our parameters to default, we utilized the BCpred epitope prediction server (http://ailab-projects1.ist.psu.edu:8080/bcpred/) by characterizing the physio-chemical features to examine the linear and continuous B-cell epitopes in the antigenic sequences (EL-Manzalawy et al., 2008). This was further validated with another B-cell epitope prediction tool called BepiPred (http://www.cbs.dtu.dk/services/Bepipred/) which was utilized to examine the epitope most likely to stimulate antigen-antibody interactions (Jespersen et al., 2017). However, from the results of the BepiPred tool, the epitope of interest was determined to be the peptides with the greatest percentile rank of 75% specificity, and those with high intensity gradients were selected.
The construction of a multi-epitope antigenic structure and the improvement of its sensitivity were both examined utilizing various online biological computing tools. Additionally, predicted were the primary, secondary, and tertiary protein structures. Prior to the construction of 3-dimension multiepitope design, an epitope with the higher B-cell prediction score was linked by an appropriate connector (GGSSGG) as shown in Figure 3A, and submitted to the I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/output/S566518/) (Roy et al., 2010). These strategies involving connection of epitopes with flexible linker sequences (Gly-Gly-Ser-Ser-Gly-Gly) can be used to create a more effective diagnostic kit (Zhang, 2008).
The backtranseq EMBOSS 6.0.1 program was used to translate the constructed B-cell epitopes into a sequence of nucleotides, from which the GC content ratio was determined. (https://www.ebi.ac.uk/Tools/st/embossbacktranseq/). Backtranslation is the process of decoding a sequence of amino acids into the corresponding codons. All synthetic gene design systems include a backtranslation module. The degeneracy of the genetic code makes backtranslation potentially ambiguous since most amino acids are encoded by multiple codons (Oladipo et al., 2022).
Codon optimization tools are online bioinformatics resources that determine the gene expression level of the multiple-epitopes in the vectors and consequently, they enhance the optimal expression of a foreign gene (multiepitope) (Olatunde et al., 2022; Ebrahimi et al., 2020). Because of the shape of the hairpin structure, higher quantities of GC sequences in messenger RNA (mRNA) can inhibit protein translation, boosting the stability of mRNA and considerably raising immunological responses to the designed diagnostic kit (Grote et al., 2005). A codon-adaptation tool-server (http://www.jcat.de/start.jsp) (Geourjon and Deleage, 1995) has been used to optimize constructs with GC content values between 30% and 70%. The back translation was likewise effective due to the insertion of the initiation codon AUG and the termination codon UAA (Oladipo et al., 2022).
The primary sequence was subjected to I-TASSER to build the tertiary structure of the multi-epitope peptide (Roy et al., 2010). In order to evaluate the secondary structure of the multi-epitope, a self-optimized prediction method using the SOPMA server (https://npsa-prabi.ibcp.fr/cgi-bin/secpred sopma.pl) was employed (Geourjon and Deleage, 1995). This approach analyzes the number of turns, loops, sheets, and helix in the protein’s secondary structure. The tertiary structure of the construct was predicted by the I-TASSER server online prediction tool (http://zhanglab.ccmb.med.umich.edu/I-TASSER) (Roy et al., 2010). I-TASSER is one of the most used tools for predicting protein structure (Iterative Threading Assembly Refinement). This server is an integrated platform that uses cutting-edge algorithms to deliver the most precise structure and function forecasts imaginable (Ebrahimi et al., 2020). The I-TASSER server also functions as an integrated platform for automated protein structure and function prediction, based on the sequence-to-structure-to-function paradigm (Roy et al., 2010).
The ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php) was used to confirm and improve the predicted structures by calculating the Z-score thus, helping to identify any errors (Wiederstein and Sippl, 2007) in the predicted structure. Assessing a protein’s 3-dimensional structure is necessary for understanding its functions and property modification (Wiederstein and Sippl, 2007). Finally, the B-cell construct was studied using the Ramachandran plot server (https://zlab.umassmed.edu/bu/rama/). The main-chain conformational propensity of an amino acid is depicted graphically by a Ramachandran plot (Anderson et al., 2005).
All of the protein sequences from the NCBI’s Protein Database were retrieved for penicillin-binding protein 2a (PBP2A). Each protein sequence was identified and recovered in FASTA format with its corresponding accession number (shown in Table 1).
Antigenic result of the retrieved penicillin-binding protein 2a
All the protein sequences exhibited a potent antigenic score at ≥0.5. These properties confirmed the suitability of the protein sequences in designing the intended diagnostic kit (Doytchinova and Flower, 2007a). The antigenicity scores are presented in Table 1.
The TMHMM server was used to screen all the protein sequences retrieved from NCBI at a scale of >0.4 and then subjected to signal peptide prediction using the SignalP server (Table 2) (Möller et al., 2001; Khazaei and Moghadamizad, 2022; Shams et al., 2021). Observation shows that all the sequences possessed three different dimensions, which implies all the proteins selected have inside, outside, and transmembrane helix positions (Table 2). Table 3 displays the outcome of the signal peptide prediction for the protein.
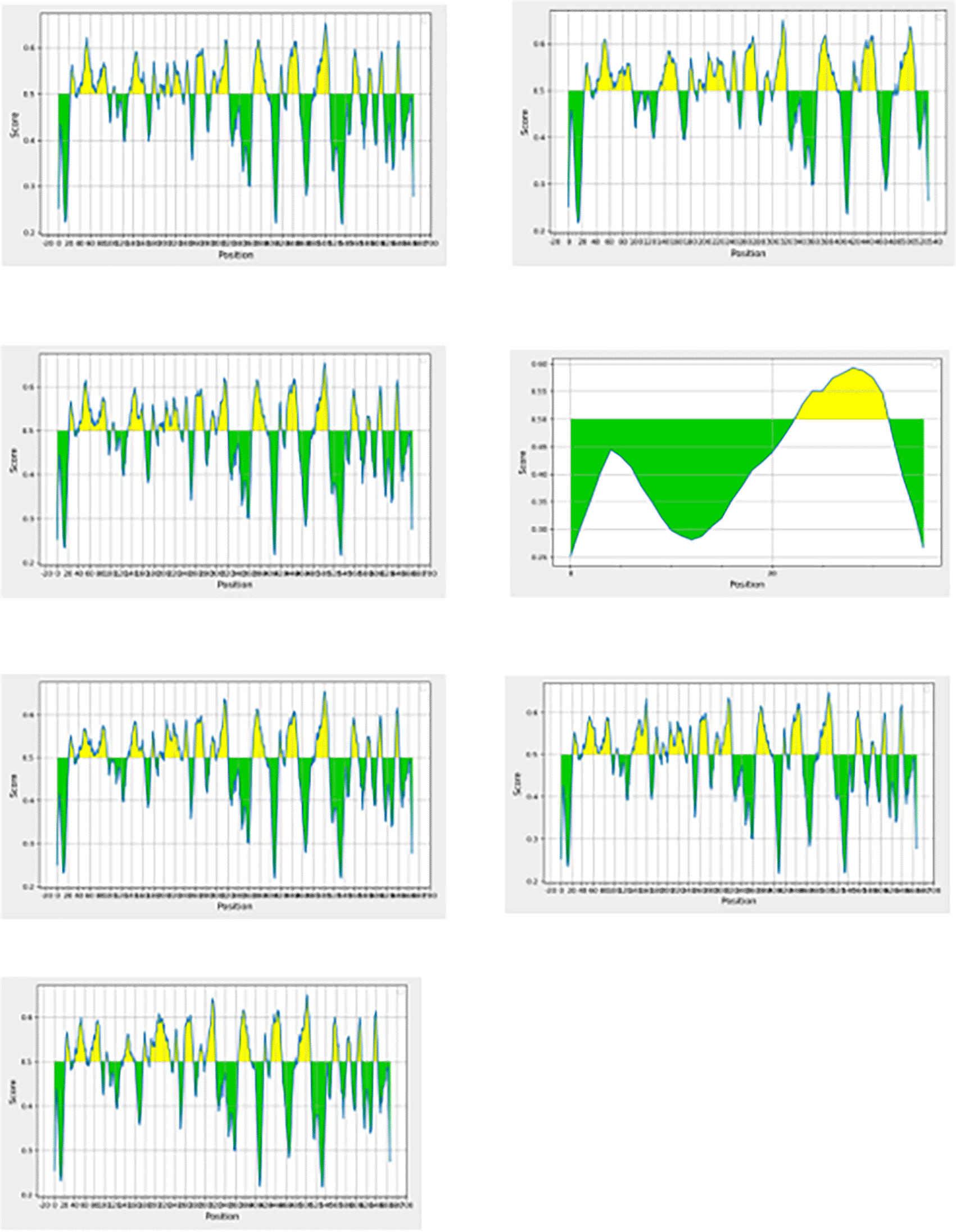
Utilizing different appropriate parameters and bioinformatics software, the predicted linear B-cells of the seven proteins are presented in Figure 2 (EL-Manzalawy et al., 2008). Altogether, 19 consented epitopes were selected for the in-silico succession; three consented epitopes from ALJ10988.1, three from ORN56903.1, three from AFJ06714.1, one from AEO00772.1, three from WP_000721309.1, three from WP_057521704.1, and three from WP_063851348.1.
Design and modelling of the multi-epitope construct
Table 4 displays a combination of consented results for the prediction of antigenic epitopes using various parameters and bioinformatics software. Each epitope was connected via a flexible linker sequence (Gly-Gly-Ser-Ser-Gly-Gly) (Ghulam et al., 2022).
Reverse translation and codon optimization
The multi-epitope protein sequences were translated into nucleotide sequences using the backtranseq algorithm. There was an optimization in the numbers of the C-G sequence. Considering the pre-optimization values of codon adaptation index (CAI) (0.92) and C-G percentage (66), there was an optimization in the numbers of the CAI and the C-G sequence following the post optimization values of 0.96 and 68% respectively. We explored the process of codon optimization in other to boost the expression of the MRSA diagnostic candidate. The results of this research are consistent with the claims that enhanced expression requires a C-G concentration of 30% to 70% and that CAI values of more than 0.8 are considered to be excellent for target organism expression (Sanami et al., 2021). The immunological response to a diagnostic kit is therefore a necessary factor that can considerably improve and increase the stability of mRNA (Ebrahimi et al., 2020).
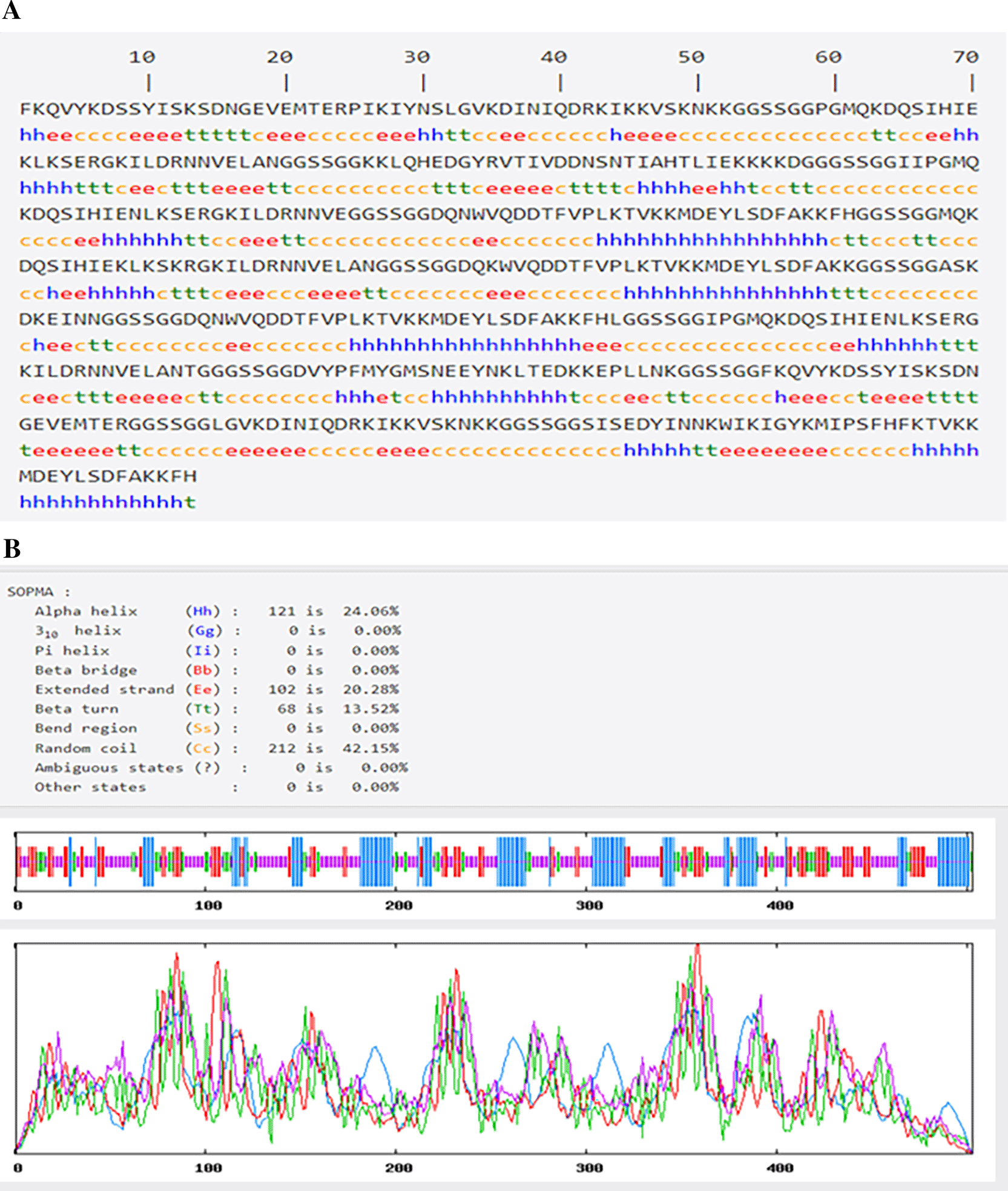
The results from the B cell primary construct with ExPASy server, containing an average buildup length of 503 amino acids, are summarized in the Table 5. To evaluate the solubility in an electric field, the theoretical PIs for the B cell construct were calculated to be 9.28. This structure is stable at a wide range of temperatures because of its 62.72 aliphatic index. The index of instability (41.44) additionally provides an estimate of protein stability in vitro, and the results demonstrate that the structures are averagely stable. Finally, the structure’s GRAVY (Grand Average of Hydropathicity) value was negative (-0.938).
The SOPMA server results are shown in Figure 3A and B. According to the findings, secondary structures that make up Alpha helixes, Beta turns, Random coil, and Extended strands are, respectively, 24.06%, 13.52%, 42.15%, and 20.28%. The prevalence of random coils and extension strands in the structure of B cells demonstrate how proteins might create antigenic epitopes.
The B-cell epitopes tertiary structure construct was predicted with the I-TASSER servers and are shown in Figure 4. For each of the targets, I-TASSER simulations provide a vast ensemble of structural conformations, known as decoys. I-TASSER uses the SPICKER program to cluster all the decoys based on pair-wise structural similarity, and then reports up to five models that correspond to the five biggest structure clusters in order to choose the final models (Kimmig et al., 2021). The relevance of threading template alignments and the convergence parameters of the structure assembly simulations are used to calculate the C-score, which provides a quantitative assessment of each model’s confidence. The values of the C-scores for models 1 through 5 are -1.50, -1.70, -1.66, -1.55, and -3.05 respectively. Model 1 with a C-score value of -1.50 was chosen because the C-score of a model typically ranges from -5 to 2, and a higher C-score number indicates that the model has a high level of confidence (Sanami et al., 2021).

A (Model 1): C-score = -1.50, B (Model 2): C-score = -1.70, C (Model 3): C-score = -1.66, D (Model 4): C-score = -1.55, E (Model 5): = -3.05.
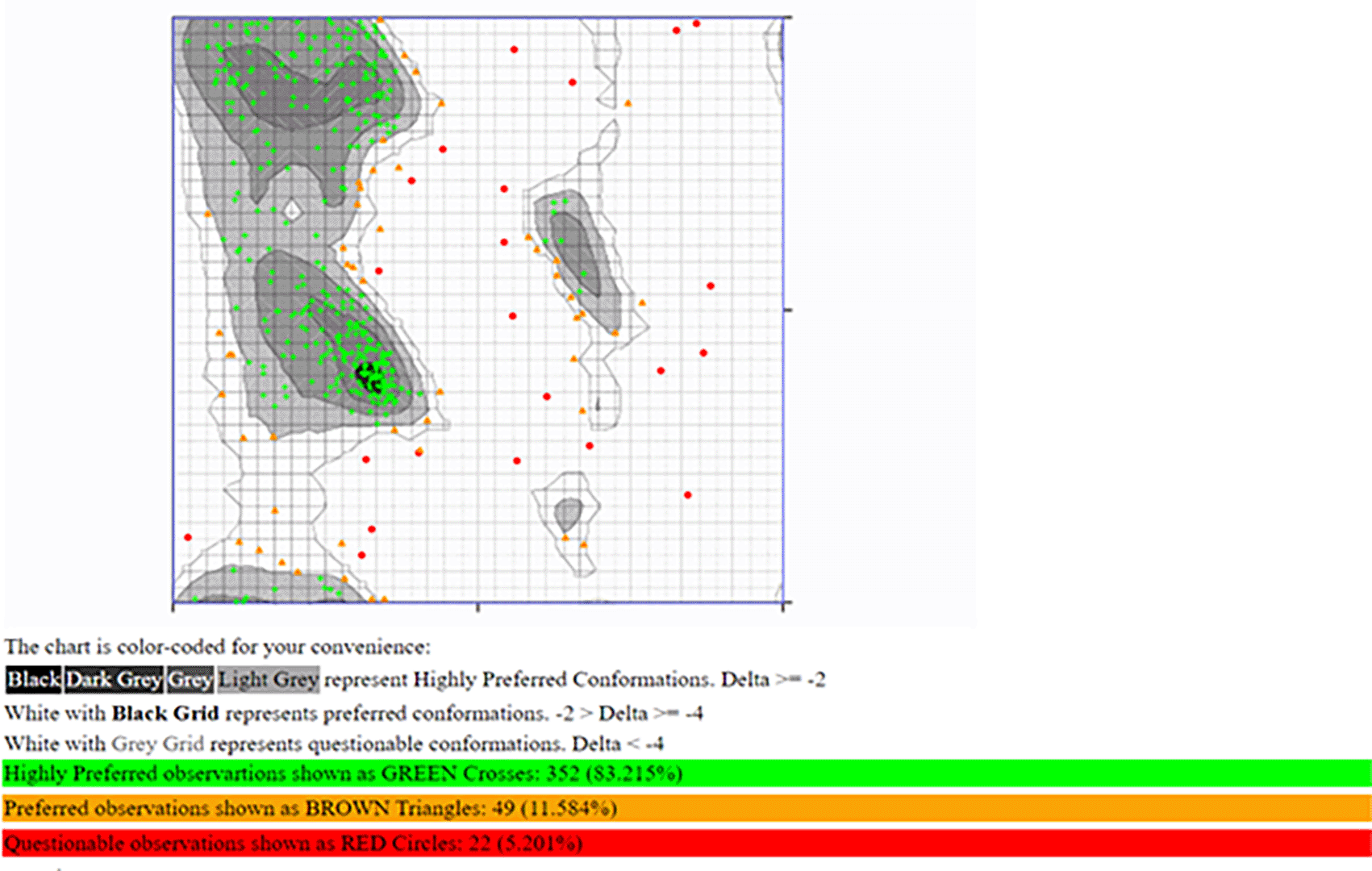
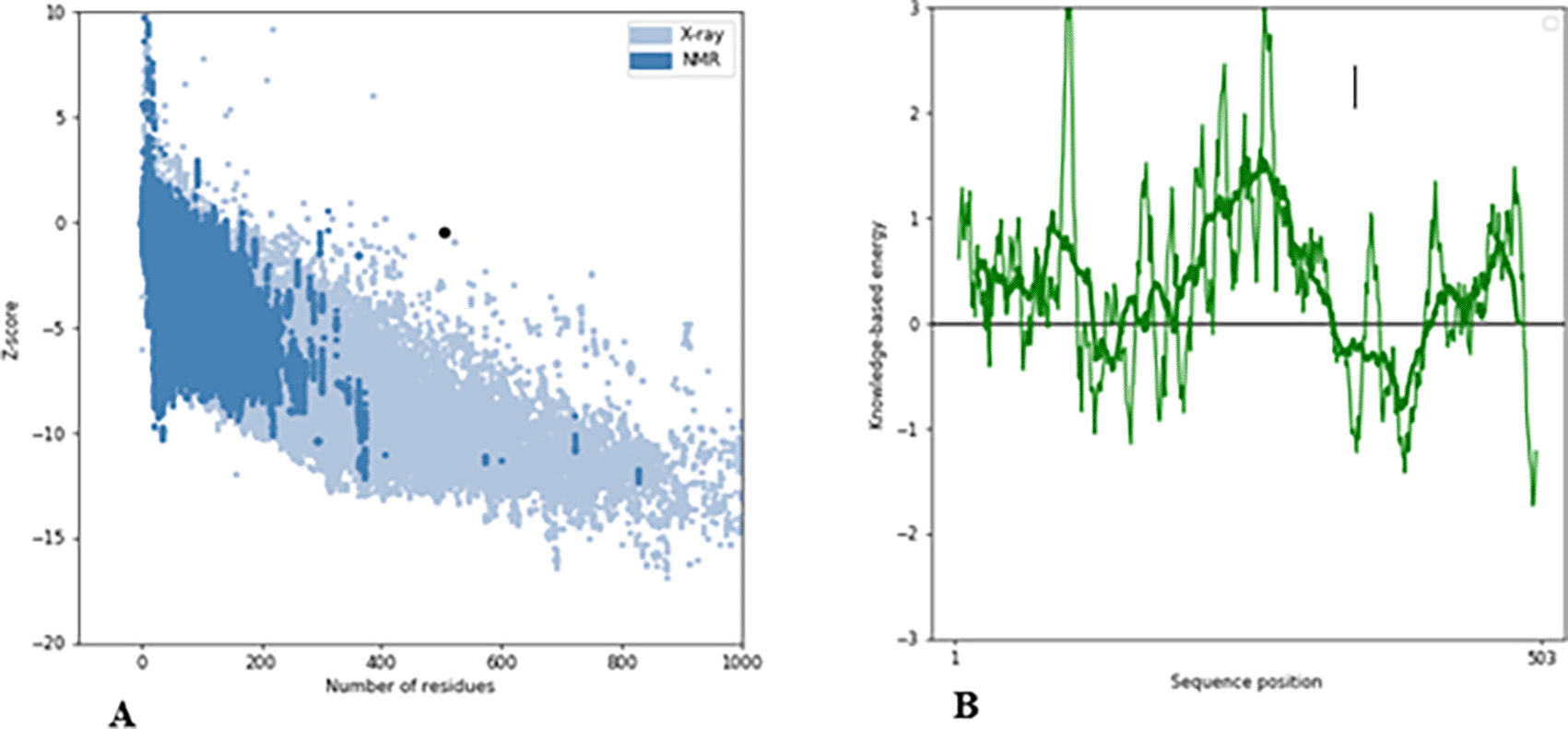
The RAMACHANDRAN PLOT server assessed the modeled structure and produced a Ramachandran plot (Figure 5). According to the results, 83.215% of the residues was discovered in the highly preferred observation, 11.584% in the preferred observation, and 5.201% in the questionable observation. Additionally, the ProSA-web server predicted both the overall model quality (Figure 6A) and local model quality (Figure 6B) with a Z-Score of -0.43 for the 3D structure of B-cell construct. These results demonstrate the 3D model of the structure’s outstanding dependability.
Methicillin-resistant Staphylococcus aureus (MRSA) is a significant global source of bacteremia in BSI. These infections are associated with morbidity and excessive mortality. The development of recommendations for the clinical diagnosis of MRSA carriage and infection is fraught with controversies (Kimmig et al., 2021; Hajissa et al., 2015). Among these are the best test methodologies, whether screening should be general or targeted, and the financial repercussions of choosing one strategy over another. It is obvious that there are significant differences in MRSA incidence and health-economic cost not only between different nations, but also between various institutions within a same country and between various patient populations. These variations will have an impact on the selection of the best screening techniques to use at both the national and local levels. What is best for patient safety, however, might not necessarily be practical financially (CDC, 2019).
Recent advancements in molecular and nonmolecular testing technologies have significantly reduced the amount of time necessary to detect MRSA. The traditional MRSA culturing and susceptibility testing takes 48 to 72 hours, including 16 to 24 hours incubation period and another 16 to 24 hours to perform the susceptibility assays. The use of chromogenic agar, which causes a color reaction in bacterial cultures, is an adaptation to the traditional culturing technique, antibiotics are also present in these medium, allowing only resistant bacteria to proliferate (CDC, 2019).
The mecA gene, which is unique to MRSA, remains a major target of amplification during PCR methods (Milheiriço et al., 2007). These tests can be directly performed on blood, nasal, or wound swab samples and typically yield results within 1 to 3 hours (CDC, 2019; Milheiriço et al., 2007). However, due to the necessity of sample transport, testing, and result communication in clinical settings, the entire time from sample collection to receiving results is frequently longer. Another method for MRSA detection is immunochromatographic assays, which utilize antibodies that react with specific bacterial proteins. If the targeted protein is present in the sample, a visible reaction occurs in the test medium. This method was described by Shin et al. (2010).
Antigen-based immunoassays are increasingly necessary at the point of care for infection detection. In-silico analysis approaches are required to evaluate the structure and functional properties of proteins. These methods could help identify antigenic regions for the development of a multi-epitope diagnostic test kit. Similarly, because of advances in bioinformatics research, epitope prediction has increased considerably in recent years (Sanches et al., 2021; Combet et al., 2000).
A diagnostic kit can be used to detect MRSA specific antibodies in serum samples (Oladipo et al., 2022; Metzger and Mordmüller, 2014). Multi-epitope peptides may be used to improve diagnostic kit accuracy, and antigen-based immunoassays are required for rapid point-of-care detection of MRSA. However, limited biomarker-based diagnostic techniques for MRSA have made it to the point-of-care (Akue et al., 2018; Metzger and Mordmüller, 2014). Consequently, this study sought to develop a diagnostic multi-epitope construct that is as accurate, specific, and efficient as possible. Using a range of immunoinformatics tools, we identified seven unique proteins with potential value as biomarkers of MRSA infection. The antigenicity of the retrieved proteins was evaluated at a threshold of 0.5, suggesting that proteins with this threshold (antigenicity score) or higher are more likely to respond to the presence of their specific antibody, as previously described by Doytchinova and Flower (2007a).
The newly designated proteins were then examined using the Transmembrane Helices Hidden Markov Model (TMHMM) server (Möller et al., 2001) to determine whether they were membrane proteins or intracellular proteins, this unique trait remains an important factor to consider when designing an antibody-based diagnostic kits. The prediction of transmembrane (TM) helices plays a crucial role when studying membrane proteins, because of the relatively small number (∼0.5% of the PDB) of high-resolution structures for such proteins (Sanches et al., 2021). Furthermore, these proteins were subjected to SIGNALP server to predict the presence and location peptide cleavage sites in the amino acid sequence. Therefore, knowing a protein’s particular signal peptide can help determine where it is most likely to be found. These analyses confirmed that the protein sequences chosen are present within plasma membrane, therefore, BcPred tools were used to identify specific epitopes within each protein sequence at a threshold of 0.5 with a specificity of 75%. According to Kringelum et al. (2012), these two tools were utilized to boost the likelihood of selecting the appropriate epitopes that can induce an immune response.
B-cell epitopes are classified into two categories; linear and conformational. Linear epitopes are continuous amino acid segments in the protein sequence while conformational epitopes are made up of amino acid residues that are physically aligned while being separated in the protein main sequence (Sanchez-Trincado et al., 2017; El-Manzalawy et al., 2017). According to our results in Figure 2, the BepiPred server predicted several linear B-cell epitopes. B-cells are responsible for the conservation of antibody while stimulating a swift response to subsequent antigen exposure (Olatunde et al., 2022; Schneider, 2017). B-cells recognize solvent-exposed antigens through B-cell receptors (BCR) called membrane-bound immunoglobulins (Olatunde et al., 2022; Schneider, 2017). Therefore, to improve the specificity and sensitivity of diagnosing MRSA infected patients, the indicated epitopes were all linked together by a flexible appropriate linker (GGSSGG) at the end of each epitope (Oladipo et al., 2022).
Further investigation of the multi-epitope construct’s primary and secondary structures revealed both the physicochemical and structural features. It was noted that the molecular weight of the designed kit is significantly greater than 32kD which is an average molecular weight for a multi-subunit diagnostic kit, thus, favoring the antigenicity of the constructs (Onile et al., 2020). The theoretical pI of 9.28 indicates that the construct is slightly basic; the pH level at which a molecule carries no electrical charge is known as the isoelectric point (pI). The idea is crucial for zwitterionic compounds like proteins, peptides, and amino acids. According to Schneider (2017), the physiological pH of human body ranges between 7.35 to 7.45, therapeutic antibodies administered with pI values between 8 and 9 are properly absorbed. Nevertheless, antibodies occasionally have pI values that fall above this range or have been altered to produce higher or lower pI values (cationization or anionization, respectively). The observed elevated aliphatic index score of 62.72 indicates that the construct is thermostable when compared to some previously developed vaccines as suggested by Onile et al. (2020). Additionally, the construct’s overall hydropathicity has a greater negative score of 0.938, indicating that it is hydrophilic. Sanches et al. (2021) established that hydrophilic properties can decrease the likelihood of functional antigen synthesis and their capacity to enhance antigen processing and presentation.
The highly precise homology modelling (I-TASSER) was utilized to obtain the 3-dimensional structure with sufficient details regarding the arrangement of protein amino acid residues and the percentage disorder. A benchmarked scoring system that enables quantitative evaluations of the I-TASSER models was developed to facilitate the creation of the I-TASSER server, which generated automatic full-length 3D protein structure predictions in Figure 4 (Roy et al., 2010; Zhang, 2008). The C-score values of the models 1-5 were -1.50, -1.70, -1.66, -1.55, -3.05, respectively. Model 1 with a value of -1.50 was chosen because the C-score of a model is typically between -5 and -2, and a higher C-score number denotes a high level of confidence Yang and Zhang (2015). Using the EMBOSS transeq server, the amino acid of the multi-epitope sequence was reverse-translated into a nucleotide sequence to ascertain the epitope’s expression level and it’s compatibility with a cloning vector. Further analysis of the designed kit’s epitopes showed that the construct’s codon adaptation index was 0.96, which was near to a score of 1.0, indicating that the construct was well expressed in a cloning vector (Grote et al., 2005).
An effective and straightforward method to assess the tertiary structure quality is to use a Ramachandran plot. Our model had 83.215% of residues in the most preferred region, while 16.785% of the remaining residues were deposited in the additional authorized regions, demonstrating the excellent quality of our model (Pande et al., 2021, 2022). Additionally, our model has a Z-score of -0.43, which indicates the overall model quality; models with lower Z-scores were thought to be of greater quality, a positive Z-score often denotes the discovery of an incorrect or inconsistent region in the created 3D protein model (Pande et al., 2021, 2022). The Z-score obtained (Figure 6) validated the high quality of our overall model (Hajissa et al., 2015; Pande et al., 2022; Olatunde et al., 2022). The major aim of developing this multi-epitope construct is to investigate their potential in the diagnosis of MRSA infection. The synthetic recombinant multi-epitope peptide employed in current diagnostic kits has proven to be sufficient and useful for designing a more sensitive and specific diagnostic test than a single epitope peptide. As a result, multi-epitope peptides are promising diagnostic markers with the potential to improve diagnostic kit accuracy.
Methicillin-resistant Staphylococcus aureus (MRSA) is a significant global source of bacteremia in BSI. Recent advancements in molecular testing technologies have significantly reduced the amount of time necessary to detect MRSA. Several in-silico approaches have been used to predict and design multi-epitope peptides capable of bounding to major histocompatibility complexes (MHC). Antigen-based immunoassays are increasingly necessary at the point of care for infection detection. In the present study, we have developed a novel multi-epitope construct for diagnosis of MRSA infection through an in-silico approach. Furthermore, the study provides an in-silico B-cell epitope mapping analysis for seven peptides, namely: ALJ10988.1, ORN56903.1, AFJ06714.1, AEO00772.1, WP_000721309.1, WP_057521704.1, and WP_063851348.1. The peptide constructs were analyzed for antigenicity, secondary and primary protein structures and physicochemical properties; investigations revealed that the peptide contains potential epitopes, hence, it can serve as a theoretical foundation for further research into its antigenicity as well as a prototype for developing a rapid diagnostic test kit for MRSA infection.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)