Keywords
In-silico drug prediction, variant prioritization, clinical relevance, CIViC database, API query
This article is included in the Genomics and Genetics gateway.
This article is included in the Bioinformatics in Cancer Research collection.
In-silico drug prediction, variant prioritization, clinical relevance, CIViC database, API query
In recent years, next-generation sequencing (NGS) has become one of the main technologies to profile the genetic landscape of tumors, offering unprecedented insights into disease mechanisms, personalized patient care and potential treatment options.1,2 One key aspect in precision oncology is the evaluation of actionable molecular alterations from cancer samples, in order to select promising targeted therapies and to predict the specific response (i.e., beneficial or adverse) of patients to a particular choice of treatment.2 However, the implementation of tailored strategies in routine cancer patient care still remains a challenging task. The wealth of data generated in standard NGS experiments, such as variant calling from whole exome sequencing (WES) or gene expression levels based on bulk RNA sequencing, needs to be interpreted in a meaningful way in order to guide clinical decision-making. Furthermore, clinical interpretation of the observed molecular profile requires an in-depth evaluation of the ever-growing biomedical literature, which is both a time-consuming and complex process that needs to be performed by experts.1,2 Altogether, a manual annotation of the oftentimes hundreds of readouts resulting from high-throughput technologies is challenging due to the amount of curation burden involved.
To overcome this bottleneck, sophisticated databases have evolved to aid the extraction of clinically relevant and actionable insights from the molecular composition of tumor samples, by enriching the identified aberrations with information such as prognosis or treatment relevance.1 Among those databases, a very popular and highly curated one is the CIViC knowledgebase, a powerful resource for the clinical interpretation of variants in precision oncology.3 This database contains expert-reviewed information about the clinical actionability of cancer genes and their molecular alterations, linking them to disease-specific knowledge about their potential therapeutic, prognostic, predisposing and diagnostic value. CIViC also provides a public application programming interface (API), which allows users to programmatically access and retrieve data from the knowledgebase.3 Nevertheless, sophisticated query tools are still required, on the one hand to enable the efficient simultaneous query of hundreds of variants, which is necessary for analyzing multiple patients in parallel. On the other hand, downstream annotation, prioritization, and summary of CIViC records is still necessary to streamline clinical interpretation. Recently, a Python package called CIViCpy has become available that offers a solution for the first issue of large-scale retrieval and inspection of CIViC records.4 This tool ensures the success of high-throughput queries by leveraging an offline version of the online content that is hosted in the knowledgebase, and it also provides valuable functionality such as coordinate search methods for the precached variants.
Despite these advancements, matching CIViC evidence to observed tumor aberrations in an automated fashion continues to be a challenge. The lookup strategies supported by CIViCpy impose limitations on the type of alterations and attributes that can be found. Moreover, the queries are exclusively coordinate-based, which can be too sensitive in case a particular amino acid change is under consideration; or it can be too restrictive, e.g. in case generally the variants affecting a particular gene are in the focus. For instance, users may wish to fetch evidence from gene expression records (which are coordinate-independent in the database), match variants on the basis of their effect in the downstream proteins rather than their genomic coordinates, or perform position-independent searches for copy number alterations in a gene. Moreover, taking full advantage of the different information available for clinical evidence in CIViC requires intricate prioritization, grouping and filtering of the extracted variant and drug information, which is not supported by CIViCpy. To this end, we implemented CIViCutils, an open-source Python package for rapid retrieval, matching and downstream processing of expert-curated evidence records from CIViC. CIViCutils can be easily incorporated into precision oncology workflows to provide variant-level disease-specific information about treatment response, pathogenesis, diagnosis, and prognosis of genomic aberrations, as well as differentially expressed genes. Convenient features offered by our package include simplified position-independent variant retrieval, subsequent match quality evaluation and prioritization based on cancer types of interest, flexible record filtering, grouping of the extracted experimental findings, and standardized reporting of the final annotations. CIViCutils is intended to facilitate the analysis and interpretation of CIViC information, with particular focus on the context of in-silico drug candidate prediction, enabling custom support during the clinical decision-making process, and in turn contributing to faster analysis turn-around times.
The package has already been applied in previous studies and analysis workflows,5–7 one of which is the automated annotation of cancer aberrations using WES variant calling data derived from the muscle-invasive bladder cancer cohort of The Cancer Genome Atlas (TCGA-BLCA).8 We use this study to showcase the functionality and use cases of CIViCutils.
CIViCutils is an open-source Python package for extracting, selecting, filtering, prioritizing, grouping and reporting variant-specific clinical information from the expert-curated knowledgebase CIViC3 (see Figure 1). It is primarily intended to be used for supplying clinical annotations to variants and drug pairs. In the following, we provide a basic overview on design choices and output. For detailed information about specific modules, required input files, and source code of CIViCutils we refer to our GitHub repository (see Software availability).
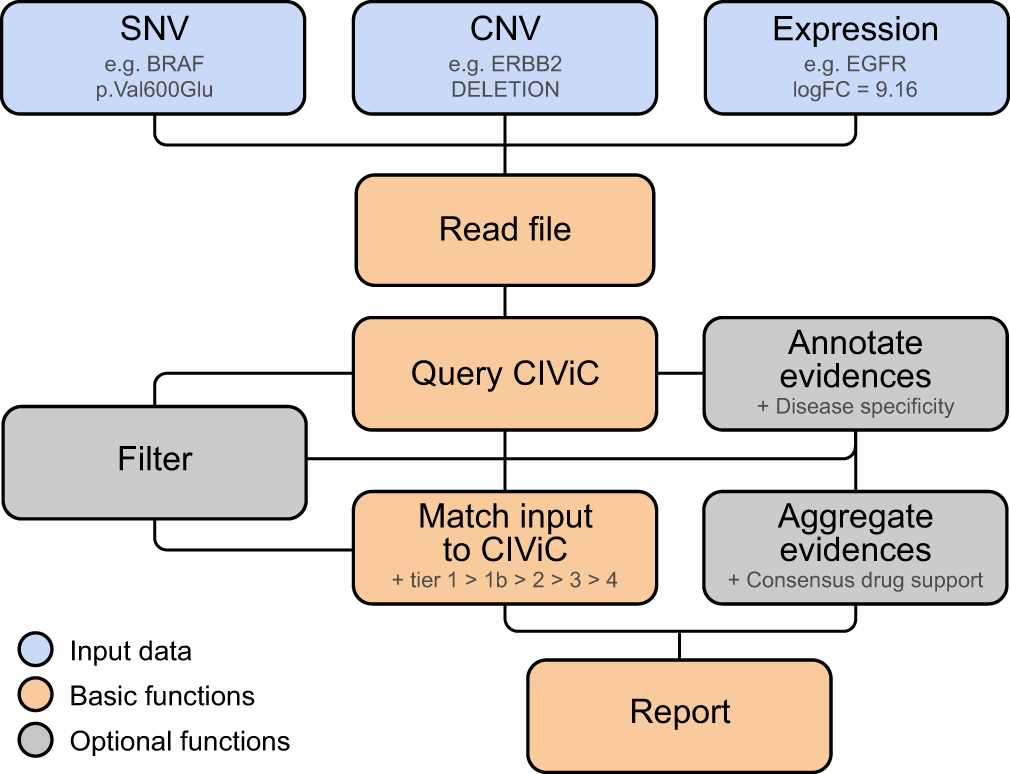
CIViCutils supports as input variant-calling data (SNVs, InDels, and CNVs) and expression data. Note that SNVs and InDels can be processed simultaneously and thus are regarded as a single category “SNV”. After the query of the CIViC knowledgebase, CIViCutils performs variant-specific matching of the provided variants to clinical evidence extracted from the database. A tier-based rating system is used for evaluating the quality of the resulting matches. In addition, the package offers functionality for annotating, aggregating, and filtering the retrieved evidences. Given one or more cancer indications that are of interest to the user, CIViCutils can further annotate data matched from CIViC with labels describing the disease specificity of the evidence. Drug prediction evidences can be aggregated (together with the cancer specificity information) into consensus drug responses. Abbreviations: SNV, single nucleotide variant; InDel, insertion-deletion mutation; CNV, copy number variant; CIViC, Clinical Interpretations for Variants in Cancer.
Input files and CIViC query
The input for CIViCutils is a list of the genes and their molecular alterations that should be queried in CIViC. The package can handle four different types of information: genomic-based data in the form of single nucleotide variants (SNVs), short insertions and deletions (InDels), and copy number variants (CNVs), as well as gene expression data from differential expression analyses. In the context of CIViCutils, SNVs and InDels are handled together and thus considered as a single category “SNV” (Figure 1). The minimum information required for a CIViCutils query are the gene names, where the specific format and content of the input file depends on the data type at hand and is described in the GitHub repository (see Software availability).
CIViCutils depends on the Python package CIViCpy4 for performing large-volume queries to CIViC, as it leverages its offline access to the knowledgebase to ease the retrieval of the often hundreds of variant records returned from high-throughput queries in standard high-throughput experiments. The query supports three different types of gene identifiers (Entrez symbols, Entrez IDs and internal CIViC IDs), and alternative gene symbols such as aliases or synonyms are also permitted during the search.
Tier-based matching of variants
One core functionality of CIViCutils is its matching framework, which associates specific variants retrieved from CIViC with the input aberrations provided by the user (Figure 1). This step is needed because oftentimes variants from different sources follow different nomenclatures, and in the particular case of CIViC records, they often deviate from the recommended and widely used guidelines by the Human Genome Variation Society (HGVS), and many entries do not even have HGVS expressions available. For this reason, we generate a standardized format for both the CIViC records and the input alterations, dependent on the type of variant being queried in each situation, and making use of HGVS guidelines whenever possible. As for CNVs and differential gene expression data which, to date, do not have any HGVS nomenclature available in CIViC, the matching is exclusively based on a reduced set of expressions known to be commonly used to designate this kind of molecular aberrations. Whenever additional information about the exon location and/or predicted variant impacts of the input SNVs and InDels has been supplied to CIViCutils, these annotations will also be leveraged by our package during the matching of variants. The quality of the resulting variant-specific matches between input and CIViC is assessed through a tier-based rating system (see Table 1). Note that, as a result of the matching framework, more than one CIViC record could potentially qualify and be assigned to the same queried variant.
Matches at the variant level are evaluated through a system of five tier categories, as described below. Categories are listed in descending hierarchical order, i.e. tier 1 matches are prioritized by the package over tier 3 ones. Note that tier 1b is only supported for SNVs/InDels, while tier 2 is not available for differential gene expression data. Abbr.: logFC, log fold-change; SNV, single nucleotide variant; InDel, insertion-deletion mutation; CNV, copy number variant; CIViC, Clinical Interpretations for Variants in Cancer.
| Tier | Description | Supported data types | Example input variant | Example matches from CIViC |
|---|---|---|---|---|
| 1 | Perfect match found between the input variant and CIViC records. | SNVs/InDels | BRAF:p.Val600Glu | V600E |
| CNVs | EGFR:AMP/GAIN/DUP | AMPLIFICATION, COPY NUMBER VARIATION | ||
| Expression | ALK:logFC>0 | OVEREXPRESSION, EXPRESSION | ||
| 1b | Non-perfect match, corresponding to CIViC records with descriptive variant names which are commonly used in the knowledgebase to designate unspecified sets of variants, e.g. of a particular type, associated to a given region of the gene, or found in specific genomic regions. | SNVs/InDels | NRAS:p.Ter213Cys | MUTATION |
| SMAD4:p.Glu55fs | FRAMESHIFT MUTATION, MUTATION | |||
| CTNNB1:p.Ser60Phe | EXON 3 MUTATION | |||
| 2 | Positional match where the protein position affected in the CIViC record is the same as the input variant, while the amino acid change differs between them. | SNVs/InDels | BRAF:p.Val601Ser | V601E |
| CNVs | EGFR:DEL/LOSS | EXON 4 DELETION, EXON 19 DELETION | ||
| 3 | No CIViC record matches the input variant, only the associated gene was found in CIViC. In this case, all variant records available for the corresponding gene and corresponding to the data type at hand, if any, are returned by the query. | SNVs/InDels | NF2:p.Glu106Lys | K159FS,MUTATION, Y177FS,C.1396C>T |
| CNVs | BRAF:DEL/LOSS | AMPLIFICATION | ||
| Expression | ALK:logFC<0 | OVEREXPRESSION, EXPRESSION | ||
| 4 | Query did not return any results, as neither variant nor gene was found in CIViC. | SNVs/InDels | KCNQ2 | - |
| CNVs | ||||
| Expression |
Annotating disease specificity
While the variant-specific clinical data returned by CIViCutils can often be considerable in size, as well as very diverse with regard to associated disease information, users frequently rather focus on a particular cancer type or even subtype of relevance during the annotation of their variants. To this end, CIViCutils allows for categorization and prioritization of CIViC data based on the specificity of their cancer indication compared to one or more indications of interest. Relevant keywords can be specified by the user and are used to match disease names of particular significance and simultaneously exclude undesired indications from the CIViC results. In addition, high-level disease names that occur in CIViC (e.g. cancer or solid tumor) can be specified and will serve as a “second-best” alternative during the classification whenever relevant terms are not found. CIViCutils reports records in three categories in descending hierarchical order: cancer type specific (“ct”), when the disease name matches relevant keywords, general cancer type specificity (“gt”), when the conditions for category “ct” are not fulfilled and the disease name matches unspecific high-level terms, and non-specific cancer type (“nct”), when none of the previous conditions are fulfilled.
Filtering clinical data
CIViCutils offers functionality for flexible record filtering at several levels of its annotation workflow (see Figure 1), allowing the possibility to clean-up and to prioritize data. For many purposes it is recommended to filter data retrieved from the CIViC query, e.g. to exclude records that have not yet been expert-reviewed, or to retrieve variants of a specific type such as somatic or germline. Furthermore, it is possible to prioritize and filter variants based on the tiers resulting from the matching framework of CIViCutils (e.g. to select clinical data from the best tier match available, or ignore input aberrations that could not be found in CIViC), as well as based on their annotated cancer type specificity (e.g. to retrieve evidences from the best classification found, or to focus exclusively on records associated with a particular disease of interest).
Consensus drug response predictions
CIViCutils provides a module for further processing and aggregating the predictive evidence annotated from CIViC into so-called “consensus drug responses”. Predictive data correspond to drug-variant interactions that can be used for in-silico prediction of the therapeutic response on the basis of actionable molecular targets. While CIViC contains a substantial number of these records, they can often be complex to interpret and quite diverse concerning content. For instance, even for the same aberration, a multitude of claims might exist across an extensive range of cancers, in turn involving various drug names and different clinical interpretations depending on the given indication. At the same time, the underlying evidence might greatly vary in terms of quantity and quality.
CIViCutils eases the interpretation of this multitude of records by combining them into a single and unanimous response prediction per aberration, and taking into account drug name and cancer type specificity. Clinical data is characterized in the knowledgebase by a combination of evidence direction and clinical significance terms. CIViCutils further interprets these records into a reduced set of expressions relative to the direct therapeutic prediction (“POSITIVE”, “NEGATIVE” or “UNKNOWN”).
In order to provide the consensus drug response prediction, first the CIViC information is standardized across records, followed by a majority vote of the available evidence (taking into account disease specificity). The consensus reported by CIViCutils is the drug response prediction with the highest number of occurrences across all records available for the therapy, cancer type specificity, and molecular alteration at hand, resulting in one of the following categories: “SUPPORT” (overall the evidence is considered “POSITIVE”), “RESISTANCE” (majority is “NEGATIVE”), “CONFLICT” (unresolved cases with contradicting information) and “UNKNOWN” (prevailing category is “UNKNOWN”, i.e. the predictive value is not known).
Output file
CIViCutils reports the annotated CIViC information into a new file, using the same layout as the input file of molecular alterations originally provided to the package. New columns are appended that summarize clinically relevant data from the knowledgebase, using an identical human- and machine-readable format regardless of the type of variants at hand.
For each variant provided to CIViCutils, information about the corresponding records extracted from CIViC is always reported with a single tier classification, rating the accuracy and overall quality of the match. Additional columns contain different aspects of the variant records, including their CIViC Actionability Scores, variant type classifications, and all associated clinical statements on disease diagnosis, prognosis, predisposition and predictive therapeutic response. Individual records are described by their specific combination of cancer indication, evidence direction, clinical significance and evidence level, as well as the publication reviewed by curators to endorse the claim. Publications are referenced using their citation identifiers, namely, PubMed sources and abstracts from the American Society of Clinical Oncology.
In addition, CIViCutils can aggregate clinical data of the same evidence type and from the same variant match to ease readability. In the first layer of aggregation, records assigned to the same evidence level are reported together under a single statement that lists the different supporting publications. In turn, claims describing the same type of clinical action (i.e. identical combination of direction and clinical significance) are also clustered, followed by the aggregation of evidence associated with identical disease names. Optionally, additional details about the CIViC records can be displayed, such as status in the database or confidence rating, as well as CIViCutils’ disease term information or consensus drug reports.
CIViCutils can be run on a Linux-based or MacOS system and requires Python 3.7, as well as an installation of CIViCpy (instructions are provided on the GitHub repository). Querying a total of 34,039 SNVs/InDels and CNVs called on the whole-exome sequencing data from the TCGA-BLCA cohort required a total of 100 MB memory and 56 minutes.
In the following we showcase different aspects of how CIViCutils facilitates the interpretation of molecular data. The examples are based on a previous study that analyzed somatic variants observed in the bladder cancer cohort (TCGA-BLCA) that is part of The Cancer Genome Atlas (TCGA).6,8 In this former study, CIViCutils was applied to a total of 34,039 SNVs/InDels and CNVs found across 412 bladder cancer patients, with the aim of identifying actionable aberrations and a set of the clinically most relevant genes and their corresponding therapies. CIViCutils was applied independently to the annotated variants observed in each tumor sample. The retrieved records were subsequently filtered e.g., in order to remove evidence not yet accepted in the knowledgebase, or data linked to germline variants. With CIViCutils input variants were matched to the available CIViC information on the basis of the best tier category. Next, the matched CIViC evidence was annotated with disease specificity information; “bladder” and “solid tumor” were provided as relevant and unspecific terms to the package, respectively. Based on this information, CIViCutils could further filter the annotated CIViC evidence to only select information from the highest cancer specificity found for every variant and evidence type. Subsequently, all remaining drug prediction data available for the matched variants were processed into consensus drug response predictions. As a result, all records with evidence direction “DOES NOT SUPPORT” were translated into drug response category “UNKNOWN”. Manual curation performed in Krentel et al. proved this type of evidence to have an ambiguous meaning, dependant on the specific context of the underlying data, hence making it difficult to translate into a clearly defined consequence without the review of an expert. Following the same logic, records associated with blank or null (“N/A”) values in their evidence direction and/or clinical significance were also considered to be category “UNKNOWN”.
The set of 34,039 actionable variants initially supplied to CIViCutils consisted of 13,514 SNVs/InDels (hereafter jointly referred to as “SNVs”) and 20,525 CNVs. The number of input SNVs available per patient spanned from 0 to 574 throughout the cohort, with an overall mean of 33 SNVs, whereas the average number of CNVs was 50, ranging between 0 and 243. Of those, CIViCutils matched CIViC information for 21% and 74% of the actionable SNVs and CNVs, respectively (see Figure 2A). The remaining variants were associated with genes that are not contained in CIViC, and hence were assigned tier 4 by CIViCutils. We refer to the Extended data (section 1) for information on the per-sample number of variants that could be matched to CIViC.9 On average, each SNV could be associated with two different CIViC variant records, whereas for CNVs only one hit was reported per individual alteration. However, overall more CNVs than SNVs could be matched in CIViC. This is due to the fact that CNVs can affect multiple genes (on average 220 genes per CNV for the variants identified in the TCGA-BLCA cohort) in contrast to SNVs that are associated with only one gene. Consequently, the likelihood of a given CNV having CIViC information available for at least one gene is higher than that of a SNV. We refer to Extended data section 3 for an overview of the identified evidence types (“Predictive”, “Prognostic”, “Diagnostic”, Predisposing”) that are available in CIViC for the variants called in the bladder cancer cohort.9
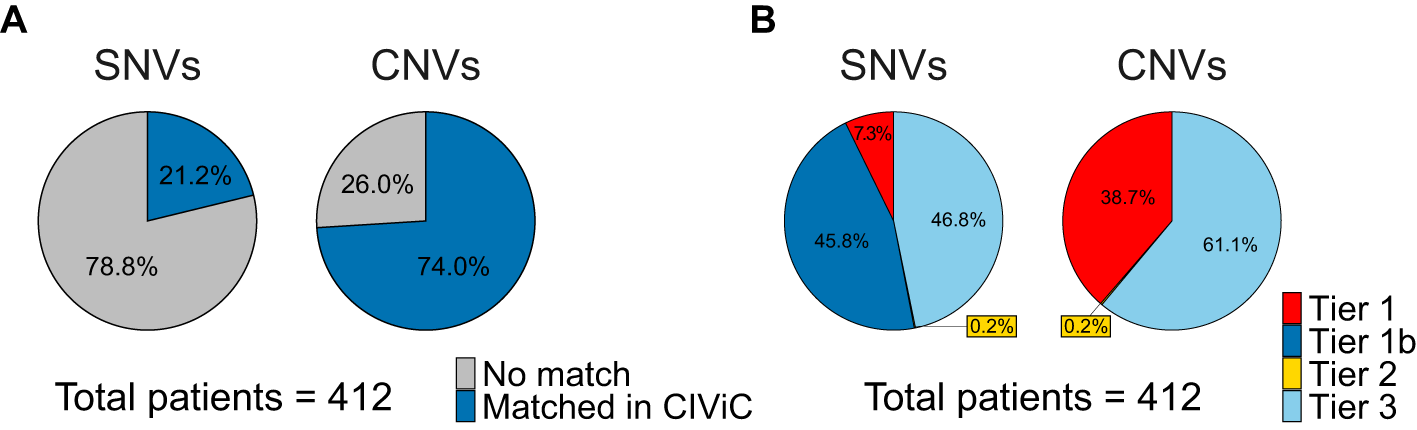
(A) Pie charts show the overall fractions of bladder cancer aberrations which were successfully matched by CIViCutils to clinical data from CIViC. (B) Pie charts illustrate the cohort-based fractions of tiers annotated by CIViCutils for the set of SNVs and CNVs successfully matched in CIViC across the 412 patients. Cancer aberrations found to have exact hits in CIViC are shown in red (tier 1), non-exact variants are represented in dark blue (tier 1b), while yellow and light blue portions illustrate positional (tier 2) and gene-only (tier 3) hits. Note that tier 1b is not available for CNVs. Abbr.: SNV, single nucleotide variant; CNV, copy number variant; CIViC, Clinical Interpretations for Variants in Cancer.
The matched records were further assigned their highest-ranking tier category (hierarchical order: tier 1 > tier 1b > tier 2 > tier 3) to assess the overall quality of the matches (see Figure 2B). Out of the 2,864 SNVs with clinical data detected across the cohort, 7% were classified as tier 1 (n=208), 46% as tier 1b (n=1,311), and 0.2% as tier 2 (n=5). From the set of 15,192 CNVs that have been successfully matched to CIViC records, 38.7% correspond to tier 1 (n=5,875), and 0.2% to tier 2 hits (n=37). On the other hand, the remaining set of tier 3 aberrations assigned by CIViCutils accounted for 47% (n=1,340) and 61% (n=9,280) of the SNV and CNV hits, respectively. Thus, in both variant types tier 3 represents the largest fraction of alterations. We refer to the Extended data (section 2) for a per-sample analysis of the tier assignment.9
Overall, exact matches were observed more frequently in the CNV set than the SNV one (39% and 7%, respectively). This is likely due to the fact that CNVs are annotated with only a few simple categories (e.g., amplification or gain) that have a higher chance to be matched compared to the complex and diverse annotations available for SNVs. On the contrary, positional matches were rarely observed regardless of the genomic alteration being considered (0.2%), in the case of CNVs, probably due to limited availability of database records fulfilling this classification, while for SNVs, it is more likely that either exact or gene-only hits were found in the database. The conditions defined for tier 1b and tier 3 are much broader and typically easier to fulfill by any variant. Accordingly, many variants match as non-exact hits, e.g., tier 3 hits represent the large majority of the retrieved CIViC matches (61%). Interestingly, tier 1b classifications (non-perfect, but of a particular type or in concrete regions of the gene, e.g., located in specific exons or introns) constitute a large proportion of matches (47%). This type of records supported by CIViCutils would not have been matched with coordinate-based searches, but it is relatively common in the CIViC knowledgebase.
CIViCutils enables the prioritization of variant matches according to disease specificity. Category ct (cancer type specific, in the TCGA-BLCA cohort analysis specified as “bladder cancer”) is the most specific match, whereas gt (general type, unspecific, in our example “solid tumor”) is the second-best match, and nct (non-cancer type specific) corresponds to cancer types differing from the cancer type of interest. Figure 3A illustrates the fraction of ct, gt, and nct matches per patient. As expected, the majority of records do not correspond to the cancer type of interest (as CIViC hosts information across many different cancer types, and only few of them match the ct term). This exemplifies the importance of an annotation of the disease specificity, as the categorization further helps to stratify the most relevant variants for each patient. We refer to the Extended data (section 4) for more information on the different disease types occurring in the nct category and more details on the observed ct and gt matches per sample.9

(A) Boxplots show the patient-based distributions of cancer type specificity labels (ct, gt, nct) reported by CIViCutils per type of genomic alteration, before and after removing tier 3 variants. Each data point (only outliers illustrated) represents the percentage of occurrences of a given disease specificity observed in one bladder cancer sample. (B) Pie charts depict the distributions of disease specificities assigned by the package throughout the TCGA-BLCA cohort, evaluated separately for SNVs and CNVs, before and after tier 3 matches were excluded. The illustrated proportions were derived from the aggregation of sample-based disease counts for every specificity label across the cohort. Abbr.: SNV, single nucleotide variant; CNV, copy number variant.
Additionally, we analyzed the overall portion of cancer indications retrieved throughout the entire TCGA-BLCA cohort per type of disease specificity and molecular aberration. Figure 3B shows for each disease specificity category the fraction of associated matches, computed per patient and aggregated across the cohort. Thus, the underlying absolute values are per category the total number of occurrences in the cohort. The vast majority of cancers retrieved by CIViCutils were labeled as nct, both in the SNV (95.4%, n=5,521) and CNV (95%, n=11,311) datasets, contrary to the remaining two categories, which overall were seldom reported and showed equivalent percentages for both types of alterations. Roughly 4% of the SNV-based (n=211) and 3% of the CNV-based (n=332) indications were annotated as ct, followed by gt, accounting for 1% (n=53) and 2% (n=266) of the extracted disease names, respectively. Figure 3A and 3B also report the percentages after removing tier 3 variants, to investigate the effect of excluding non-exact matches from the set of variants. Excluding tier 3 records has little effect on the overall results, except that for SNVs no longer the gt category can be observed.
CIViCutils generates consensus drug response predictions for variants matched to CIViC records with predictive evidence, taking into account disease specificity information. Figure 4A shows per sample the number of variants with at least one consensus prediction. On average, treatment response information was reported for 75% of the SNVs and 50% of the CNVs. The percentage of genomic alterations linked to treatment predictions increased when excluding non-exact (tier 3) matches, and is then comparable between SNVs and CNVs (on average 85% and 93%, respectively).

(A) Boxplots illustrate the percentage of variants with drug response predictions across the cohort, before and after tier 3 matches were excluded. (B) Boxplots depict the fractions of unique therapies reported for every sample, classified by their sample-level drug response derived from all the consensus predictions available in each case (“ALL-SUPPORT”, “ALL-RESISTANCE”, “ALL-CONFLICT”, “ALL-UNKNOWN”, “MIXED”). Every data point (only outliers shown) represents the fraction of treatments observed in one patient in the respective response category. Abbr.: SNV, single nucleotide variant; CNV, copy number variant.
Figure S5 (see Extended data, section 59) shows the mean number of response predictions available per variant. On average four entries were available per SNV and three entries per CNV. Per sample and treatment, different consensus prediction categories can be assigned: “ALL-SUPPORT”, “ALL-RESISTANCE”, “ALL-CONFLICT”, “ALL-UNKNOWN” and “MIXED”. In the first four categories, the treatment was consistently associated with the same drug-level prediction (e.g. “SUPPORT” for “ALL-SUPPORT”) across all the evidence records and variants observed in a sample. In the case of treatments classified as “MIXED”, different responses were reported for the same therapy and patient depending on the particular variant being evaluated. As shown in Figure 4B, the most prevalent responses assigned across TCGA-BLCA patients were “ALL-SUPPORT” (64%) and “ALL-RESISTANCE” (23%), which together accounted for over 87% of the therapies predicted on average per tumor. The high number of supporting evidence records goes in line with a known reporting bias for positive experiment results, including positive associations with treatment response.3,10 Importantly, divergent and non-informative response predictions were only rarely reported. Category “ALL-UNKNOWN” was on average annotated for only 6% and 8% of the SNV-based and CNV-based drugs, respectively, followed by “MIXED” therapies, where the mean fractions observed per patient were of 1% for the SNVs and 5% for the CNVs. Only 1% of the annotated therapies were assigned an “ALL-CONFLICT” prediction. These observations are similar when excluding non-exact (i.e., tier 3) variant matches. We refer to the Extended data (section 5) for details on the prediction types observed for individual variants.9
To allow comprehensive tumor profiling as a personalized strategy for supporting clinical decision-making in precision oncology short analysis turn-around times and simplified interpretation of the actionable molecular aberrations observed in cancer patients is required. In this context, well-curated knowledgebases such as CIViC, which link aberrations to their potential effect on prognosis and treatment response, are of high importance. Here, we introduced CIViCutils, a user-friendly and open-source Python package for the automated enrichment of tumor aberrations with CIViC information. Our package facilitates the extraction, analysis, and interpretation of expert-reviewed clinical data from the CIViC database. CIViCutils can be easily integrated into clinical workflows for comprehensive tumor profiling and it supports as input genomic aberrations (single nucleotide and insertion-deletion variants, and copy number alterations) as well as gene expression data. The package has been already employed in existing clinical analyses workflows, where it provided real-world clinical decision support.5–7 We foresee continuous package development for additional applications, such as extending the package to support queries from other variant-level clinical databases (e.g. OncoKB11 or ClinVar12).
In our use case example on analyzing actionable aberrations detected in 412 tumor samples from the TCGA-BLCA study, we show that CIViCutils could retrieve CIViC information for 21% and 74% of the actionable SNVs and CNVs, respectively. While for those records typically a wealth of clinically relevant information is available, this proportion also shows the current general limitation of relying on highly-curated knowledgebases: such high quality and expert curated information is typically not available for thousands of variants but only a subset. Nevertheless, the databases are constantly growing, leading to more frequent hits in the future. Moreover, having reliable information even for a fraction of hits greatly aids the interpretation and reduces the overall burden of prioritizing the clinically relevant results.
We highlight that using CIViCutils in the future to annotate the WES data from the TCGA-BLCA cohort would likely deliver different results than those described in our study, due to the ever-growing research literature and ongoing manual curation efforts in CIViC. Thus, the success of our package is heavily reliant on such resources becoming extended and more curated over time, with the ultimate goal of overcoming the current challenges of variant interpretation in cancer.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)