Keywords
Hypoxic-Ischemic Encephalopathy, Neurological disorders, Genetic variants, Neurotransmitters.
This article is included in the Genomics and Genetics gateway.
Hypoxic-Ischemic Encephalopathy, Neurological disorders, Genetic variants, Neurotransmitters.
Hypoxic-ischemic encephalopathy (HIE) appears in neurological conditions affecting brain areas with higher metabolic rates and active myelination processes.1 HIE is the most common cause of death and disability in neonates,2,3 causing 23% of all neonatal deaths worldwide and affecting 0.7–1.2 million infants annually.4 For instance, in developed countries, neonatal encephalopathy is 1 to 3 per 1000 live births at term, whereas low and middle income countries (LMIC) have an incidence of 1 to 5 per 1000.5,6 Moreover, its manifestation and severity are variable. Amongst those who survive the initial injury, rates of disability remain high throughout life.7 For example, in severe asphyxia, there is a 60-100% chance of having long-term sequelae; in moderate encephalopathy, there is a 20-40% chance of having significant neurological sequelae.8 The sequelae can vary, displaying sensory or cognitive abnormalities that persist to adolescence, cerebral palsy (CP), motor deficits, developmental delays, speech delays, learning disabilities, behavioral and emotional disorders, hearing impairments, and visual and feeding impairments.7,9
The disorder known as hypoxic-ischemic encephalopathy (HIE) encompasses a wide range of causes, including reduced blood flow and oxygenation to the brain before, during, or after birth. Common factors contributing to HIE include issues with the umbilical cord, uterine rupture, preeclampsia/eclampsia, placental abruption, previous placenta complications, anesthesia errors, low amniotic fluid, premature rupture of membranes, premature birth, prolonged or halted labor, excessive uterine contractions, fetal stroke, post-maturity syndrome, and delayed emergency cesarean section.10,11 Although HIE is typically associated with fetal causes, there are neurogenetic disorders that resemble HIE clinically but have distinct underlying causes, natural progressions, and prognoses. These conditions are referred to as hidden HIE states. It is crucial to remain vigilant in identifying the underlying cause, as it may involve a treatable genetic or metabolic factor. Infants who experience perinatal asphyxia, a form of HIE, may initially exhibit muscle hypotonia and later develop dyskinetic forms of cerebral palsy in the following years. This complexity further complicates the identification of hidden HIE states.12
Inborn errors of metabolism (IEM) are mainly expressed as nervous system diseases with diverse clinical characteristics and manifest as a neurodevelopmental disorders. Because IEM can present in the neonatal period with neurologic distress, metabolic acidosis, and multiorgan system involvement, they can be easily confused with typical features encountered in HIE.13 Some critical HIE conditions classified as IEM include disorders of neurotransmitter metabolism, such as nonketotic hyperglycinemia (NKH) and catecholamine metabolism disorders, which are caused by genetic changes resulting in abnormalities in the synthesis and degradation of a neurotransmitter.14 The incidence of these disorders is mainly known in first-world countries. For instance, it is known that the NKH has an incidence of 1:63,000 live births and 1:55,000 newborns in British Columbia and Finland, respectively.15 Its incidence is still being determined in developed countries like Colombia. Classically, NKH is associated with normal pregnancy, neonatal apnea, lethargy, hypotonia and seizures, and severe psychomotor retardation in those who survive.
Recent techniques such as next-generation gene sequencing (NGS) are constantly evolving and are expected to be expanded and refined in the future.16 NGS is a set of techniques that simultaneously allow rapid and accurate determination of multiple genes, providing an effective tool for diagnosing and personalized treatment of neurogenetics disorders.17 These techniques can potentially improve the quality of life of affected patients through early identification and individualized treatment of these disorders.18
NGS technologies have been put forward as tools to correlate the outcomes of patients with hypoxic-ischemic encephalopathy, as they can help detect genetic mutations in patients with hypoxic-ischemic encephalopathy that could predict the severity of brain damage and the likelihood of recovery.19,20 These diagnostic tests, such as NGS-based gene panels, can also guide treatment and early intervention.21 Consequently, NGS technologies are important tools for researching and treating hypoxic-ischemic encephalopathy, as they allow a better understanding of the molecular and genetic changes associated with brain injury. The study aimed to evaluate patients who, at birth, met the criteria for HIE to determine the origin of their clinical phenotype through the approach to personalized medicine by correlating the functional and neurodevelopmental outcomes with magnetic resonance imaging (MRI) and genetic findings.
Our study followed the “Strengthening the Reporting of Observational Studies in Epidemiology” (STROBE) statement standard checklists.
We conducted a prospective cohort study of consecutive asphyxiated newborns (n=28) admitted to the neonatal intensive care unit of several 4-level Clinics in the center-west of Colombia from January 2015 to December 2021. The determination of potentially eligible participants was based on the careful selection of a clinical team, including neonatologists, who thoroughly evaluated each newborn for the presence of HIE-related criteria and suitability for inclusion in the study. The institutional ethics committee approved all protocols and obtained parental written informed consent for each patient. Ethics Committee Review Board approved the study at COMFAMILIAR RISARALDA CLINIC (Approval No. 00049, date 2019-05-09). The newborns were selected according to the criteria of HIE indicated by the American College of Obstetricians and Gynecologists and the American Academy of Pediatrics, such as (a) umbilical cord arterial pH less than 7, (b) Apgar score between 0 to 5 for longer than 5 min; (c) neurological manifestations such as seizures, coma or hypotonia and (d) multisystem organ dysfunction (e.g., cardiovascular, gastrointestinal, hematological, pulmonary or renal system).22 In addition, the severity of its manifestation/neurological damage was evaluated according to the SARNAT staging (i.e, clinical staging of HIE),23,24 MRI assessment, and Bayley scale III.25
In this study, the Bayley Scale III was used to assess the development of the 28 patients. A certified physical therapist administered the scale (at COMFAMILIAR Risaralda, COL), which consisted of three subtests: cognitive, language (including receptive and expressive communication), and motor (including fine and gross motor). The Bayley III provides norm-referenced composite scores for each skill area, with a mean of 100 and a standard deviation of ±15. Based on the scores, the patients’ psychomotor development profiles were characterized as extremely low, borderline, low average, average, high average, superior, or very superior. Additionally, the developmental age of each patient was identified using this scale. Furthermore, the psychomotor development of the children was assessed during their first year of life at 3, 6, 9, and 12 months, respectively.
The Bayley-III Child Development Assessment Scales are a set of standardized assessment scales that allow us to evaluate the mental, psychomotor, and behavioral development of children between 1 and 42 months. These scales allow the identification of neurodevelopmental disorders and are measured according to percentiles, developmental age, and qualitative assessment comprising: extremely delayed (), borderline (70-79), below average (90-109), high average (110-119), superior (120-129) and very superior ().
Brain MRI scans were evaluated for neurological damage in 23 of 28 patients. The MRI scans were conducted during the first month of life to evaluate the severity of the lesion. Each MRI included anatomic T1- and T2-weighted imaging and diffusion-weighted imaging (DWI). MRI images were interpreted by a Senior neuroradiologists at Comfamiliar Risaralda, who were blinded to the clinical condition of the infants. Each MRI was scored using an MRI scoring system,26 consisting of a basal ganglia (BG) injury scale, a watershed (W) pattern injury scale, and a basal ganglia/watershed (BG/W) pattern injury scale. Based on the above, we classify patients into normal or abnormal MRIs.
In order to assess if the clinical phenotype of each patient is related to some genetic alteration that could be acting as an HIE masker, we used an NGS sequencing panel for the 28 patients for genes related to neurotransmitters. Additionally, we used the Microarray comparative genomic hybridization (CGH) test for 10 patients with phenotypic alterations or neurological compromise to identify the copy number imbalance of DNA (deletions and duplications) and complete exome (samples were taken at Comfamiliar Risaralda and processed by GENCEL PHARMA COL in Bogota). Hence, blood samples were collected between 2020 and 2021 from all patients and sent to an external laboratory for their analyses (DNA amplification and bioinformatic analysis). Therefore, the authors were not involved in the sequencing or variant identification. Finally, the variants were classified as I) pathogenic variant (PV), which refers to alterations with solid evidence (on databases and literature) to support that the variant is disease-causing, or II) variant of unknown significance (VUS), which refers to alterations with limited and conflicting evidence regarding pathogenicity.
A descriptive analysis was carried out on all the study variables. Categorical data analyzes were applied to nominal and ordinal variables, summarizing them as frequencies and proportions. Numerical data were summarized using mean, median, standard deviation, and interquartile ranges according to the distributional properties of these variables. In order to identify differences between patients with and without sequelae, a hypothesis contrast analysis was carried out. Since the sample sizes were small, the assumptions of normal distribution were not assumed, and nonparametric tests such as Chi-square and Fisher’s exact test were used. An analysis was conducted to examine the association between the presence of mutations and the development of sequelae. A binary logistic regression analysis was performed to adjust the odds ratio (OR) and estimate confidence intervals to assess relevant variables. The OR was considered significant if the adjustment resulted in a change of or more compared to the crude OR. The analyses were conducted using R software, and the report was generated using the gtsummary package.27
The methodology employed in this study involved a systematic and comprehensive approach, as depicted in the process flowchart in Figure 1. Firstly, a cohort study was conducted, and eligibility criteria were applied to select patients for further evaluation. The psychomotor development of the selected patients was assessed using the Bayley Scale III, allowing for the characterization of their developmental profiles. Additionally, brain MRI data were evaluated to identify any neurological damage. Genomic analysis was performed using NGS sequencing and Microarray CGH tests to explore potential genetic alterations. Finally, statistical analyses were conducted to analyze the data and investigate associations. This approach facilitated the systematic investigation of patients’ clinical profiles and genetic factors, contributing to a comprehensive understanding of the research objectives.
The study included 28 infants diagnosed with HIE who met the inclusion criteria (10 [35.7%] females; 18 [64.3%] males). The encephalopathy grade of each newborn was characterized according to the SARNAT scale, finding that the majority of patients were classified in SARNAT type 2 (n=15), followed by SARNAT type 1 (n=10) and SARNAT type 3 (n=3). In total, only 12 patients had clinical seizures at birth (confirmed by electroencephalogram). Regarding the birth characteristics, 17 patients (60.7%) come from a vaginal birth and 11 (39.3%) by cesarean section.
Table 1 shows demographic information related to the descriptive data.
According to the evaluation, it was found that 16 of the 28 patients in the study presented alterations in one or more of the scales performed, with scores <69, suggesting neurodevelopmental disorders.
The neurodevelopment evaluated in the cognitive, expressive, and receptive language, fine, and gross motor scaales presented scores between 0 and 79 in 9/28 patients, suggesting neurodevelopmental disorders. Five of these nine patients with disorders presented later diagnoses (as part of the research) of spastic cerebral palsy (a neurological condition affecting muscle control and coordination) and dyskinetic (characterized by involuntary movements). 16/28 patients presented alterations in the specifically expressive language scale with scores between 0 and 79.
These findings highlight the importance of early identification and intervention for developmental delays, as they can significantly impact a child’s long-term outcomes. Healthcare professionals must be aware of the signs and symptoms of developmental delays and refer children for appropriate assessment and support.
MRI was performed on 23 patients, with 12 showing normal results while 11 had abnormalities. It is important to note that the MRI was not performed on all patients due to a combination of factors. Some parents did not authorize the procedure, and the treating physician did not approve it unless there was a medical indication, such as in the case of patients with SARNAT 1. The patients with global developmental delay had various findings, including perirolandic cortical involvement, involvement in the basal nuclei (lenticular and thalami), basal ganglia involvement in the thalamus, central thalamic involvement, cerebral cortex involvement, involvement in the nucleus of the base, and decreased corpus callosum. They also had germinal matrix hemorrhage grade 2 on the left side, diffuse supratentorial parenchymal injury with signs of cytotoxic edema, and multiple bleeding involving different parenchymal lobes as the involvement of bilateral cerebellar parenchyma. Some patients had multiple bleeding in the cerebral parenchyma, some with liquid-liquid levels. On the other hand, patients with language delay had different findings, including affected pre- and post-central cortex and involvement of basal nuclei (lenticular and thalami). They also had a right temporal focal ischemic event, cortical edema, and bilateral Pareto-occipital lamellar subdural hematoma with extension to the tentorium. Furthermore, an alteration in the signal intensity of the basal nuclei and thalami with a restriction focus in the splenius of the corpus callosum was not associated with sequelae.
Based on NGS sequencing and Array GCH, genetic analysis of the patients was performed, where it was observed that 17 (60.7%) of them did not present any genetic alteration, while the remaining 11 (39.3%) patients presented a total of 13 genetic variants. Figure 2 shows detailed information on the genetic analysis performed on 28 patients. In addition, we can see that red borders represent pathogenic variants, blue borders represent variants of uncertain significance (VUS), and those with a light yellow filling are directly associated with the clinical diagnosis of the patients. Of these, 10 patients had single nucleotide polymorphisms for genes related to neurotransmitter disorders. Among these 10 heterozygous variants were found, three are classified as pathogenic (ALDH7A1, SLC1A4, and MYH2), and seven are variants of uncertain significance (TH, DBH, GCH1, SLC6A5, ABAT, ALDH5A1, GLRB). In addition, a pathogenic variant was found in homozygosis in the AMT gene, which is related to a cause of non-ketotic hyperglycinemia.
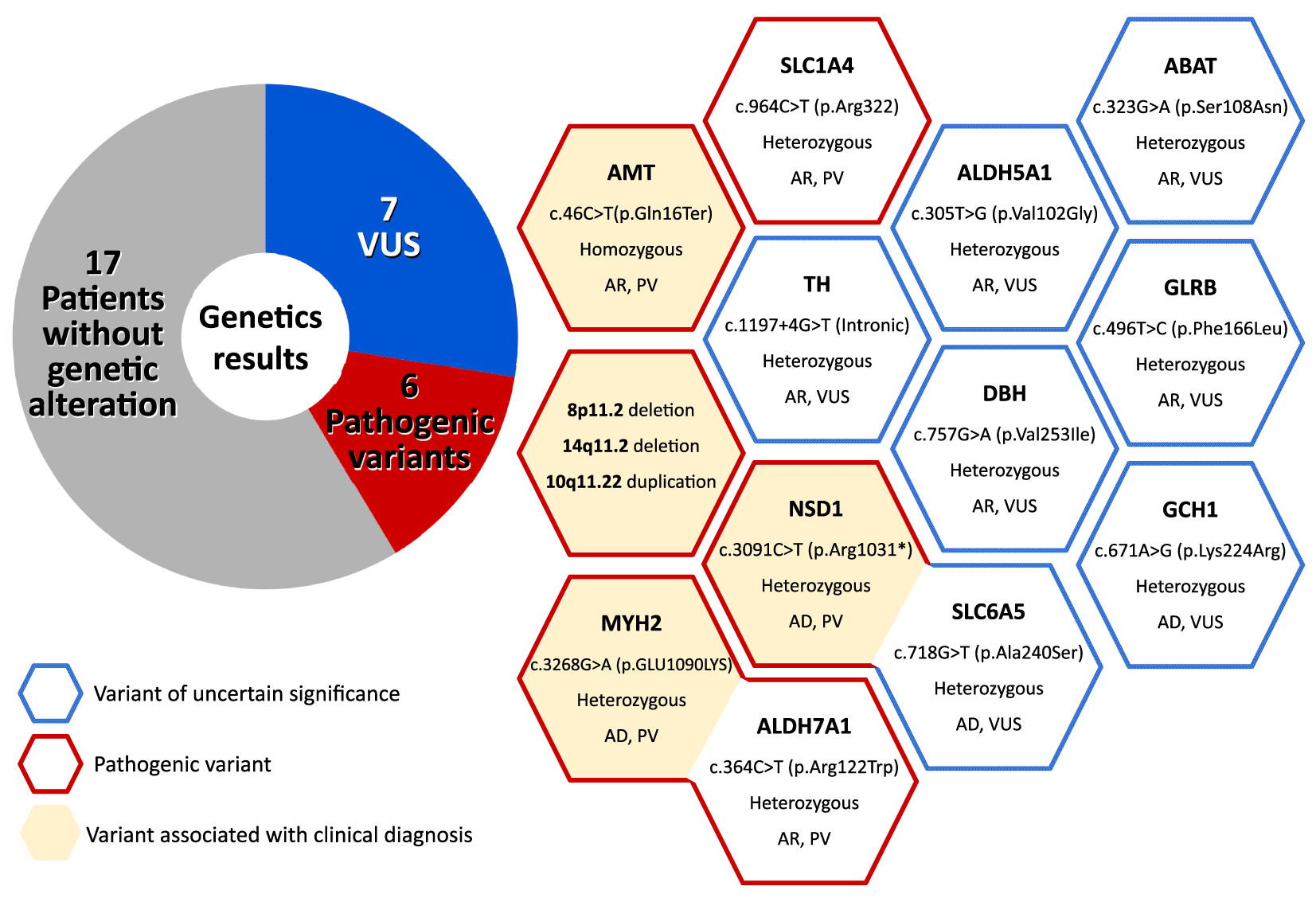
Regarding other findings, one patient presented DNA copy number imbalance, with two deletions (8p11.2 and 14q11.2) and one duplication (10q11.22), the highest classification on the SARNAT scale (type 3), and severe neurological alterations. In addition, an alteration in the NSD1 gene was identified in another patient, which is related to Sotos syndrome and is associated with delays in cognitive and motor development, as well as the presence of seizures.
The presence or absence of sequelae was analyzed in relationship with variables such as genetic mutation, type of SARNAT, seizure, and MRI scans (Figure 2, Tables 1 and 2). The qualitative analysis showed that 9 of 11 patients with a genetic mutation, either pathogenic or of uncertain significance, presented sequelae (Table 1). The only two patients who did not present this relationship were the newborns with a mutation in the ABAT and GLRB gene (both of VUS and heterozygous). Furthermore, it should be noted that these patients have normal MRI results. In addition, within the group of those who presented sequelae (n=16, 57.1%), eight newborns showed an abnormal MRI result, and five showed a normal MRI. The rest of the patients had no sequelae, even if they had experienced seizures at birth or an abnormal MRI result. This pattern is supported by analyzes of a statistical association between those clinical characteristics (SARNAT, seizures, MRI, and genetic findings) and the presence of sequelae (Table 2, which indicated that the presence or absence of mutations tended to be associated with the presence of sequelae (p-value=0.054) and a significance of p-value=0.04 for the pathogenic variants.
(VUS: Variants of unknow significance; PV: Pathogenic variants).
Finally, the logistic regression model taking as the dependent variable the presence of sequelae, the exposure variable the presence of genetic mutations and adjusted with the variables: hypothermia, seizure, altered MRI, and SARNAT (Table 3 showed a higher risk of developing sequelae when there is a genetic mutation compared to patients who do not present it (OR: 6.43; 95% CI: 1.2-55.5; p-value: 0.044). On the other hand, there is an increased tendency to develop sequelae when the MRI is altered (OR: 8.27; 95% CI: 1.39-77.2; p-value: 0.034) and when the manifestation of SARNAT is greater (OR: 8.82; 95% CI: 1.38-98.1; p-value: 0.038).
| Effect without adjustment | Adjusted effect | ||||||
|---|---|---|---|---|---|---|---|
| Characteristic | ORcr1 | 95% CI1 | p-value | Adjustment | ORaj2 | 95% CI1 | p-value |
| Genetic mutation | 6.43 | 1.2-51.5 | 0.044 | Hypothermia | 5.63 | 0.99, 46.6 | 0.068 |
| Seizure | 5.1 | 0.72, 54.9 | 0.13 | ||||
| Altered MRI | 8.27 | 1.39, 77.2 | 0.034 | ||||
| SARNAT | 8.82 | 1.38, 98.1 | 0.038 | ||||
There is no relationship concerning sequelae for the different SARNAT types and the presence/absence of seizures. However, it is noted that SARNAT type 3 manifestations coincide with the presence of sequelae. Besides, the relationship of the SARNAT score concerning the presentation of seizures, the type of MRI result, the appearance of sequelae, and the presence of genetic mutations, is presented in Figure 3. This figure presents a parallel association for all patients from a personalized medicine approach. Although the associations in Figure 3 are unique to each patient, the expanded view allows one to observe relationship patterns between groups of patients, where the blue lines represent the analysis of individual patients and the yellow lines highlight those patients who were confirmed to have a neurogenetic diagnosis.

The graph shows relationships between the following variables: SARNAT type, seizures, therapeutic hypothermia, MRI, genetic mutations and sequelae.
This study evaluated patients who met the criteria for HIE at birth to determine the origin of their clinical phenotype through the approach to personalized medicine by correlating the functional and neurodevelopmental outcomes with MRI and genetic findings. By doing so, we studied the underlying mechanisms of HIE and identified potential targets for personalized interventions that can improve patient outcomes. Some neurodevelopmental disorders that can result from perinatal asphyxia include: a) cerebral palsy: these disorders affect a person’s ability to move and maintain balance and posture; b) seizure disorders: oxygen deprivation during birth can cause damage to the brain and result in seizure disorders such as epilepsy; c) developmental delays: children who experience perinatal asphyxia may have delays in achieving developmental milestones such as walking, talking; and d) socializing, language and learning difficulties: oxygen deprivation during birth can also result in language and learning difficulties in children. Thus, the results report that vaginal childbirth was the most common among patients (60.7%). However, three patients (10.7%) showed that the other cases (89.3%) manifested themselves under birth complications, reaffirming that this is one of the factors associated with the development of PA.28
The results showed that three out of three (100%) patients had severe encephalopathy derived in long-term sequelae, and six out of 16 (37%) patients had moderate encephalopathy derived in significant neurological sequelae. As mentioned above, these sequelae were assessed using the Bayley Scale III, allowing relevant quantification of neurodevelopmental such as language, cognitive, and motor alterations. In addition, out of the 28 patients included in the study, 16 had language problems, and nine had cerebral palsy. Based on the Bayley Scale assessment, it was found that 32.14% of the patients experienced cognitive compromise. Among the patients, five of them were diagnosed with epilepsy. Treatment for these neurodevelopmental disorders varies depending on the specific disorder and its severity and may include medication, therapy, and other interventions. Early diagnosis and intervention can be especially critical in improving outcomes for children affected by PA.
Furthermore, genetic testing can also help families better understand the nature of their child’s condition and help them connect with support groups or other resources. Genetic testing can help families make informed decisions about their child’s care and treatment by providing a clearer understanding of the child’s condition. A genetic diagnosis of neurodevelopmental genes can help improve the quality of life of children by providing information about the underlying causes of their neurological or psychiatric conditions.29 Besides, this information can help doctors and families develop a more personalized and effective treatment plan for the child.
In this study, 13 genetic alterations related to neurodevelopment for 11 patients have been identified. The seventh identified has an uncertain clinical significance, while two patients are indicated as carriers of specific pathogenic mutations (ALDH7A1 and SLC1A4). The patient with a pathogenic mutation in ALDH7A1 presents an additional mutation in the MYH1 gene related to congenital Mmyopathy 6 with ophthalmoplegia matching the patient’s phenotype. In addition, another patient carrying a pathogenic mutation in heterozygosity for the SLC1A4 gene is associated with spastic tetraplegia, thin corpus callosum, and progressive microcephaly, an autosomal recessive disease. Although the patient’s phenotype coincides with this genotype, additional studies on the gene are necessary to confirm the association. Furthermore, these findings contribute to scientific knowledge and understanding of the pathogenesis of the disease, which in turn may lead to new therapeutic and preventive strategies in the future.
One patient had nonketotic hyperglycinemia with a homozygous mutation in the AMT gene. By understanding the underlying biochemistry of a child’s brain, physicians can prescribe drugs specifically designed to target the neurotransmitter systems involved in the disease.30 Therefore, implementing accurate, individualized management with a glycine-free diet can reduce symptoms and improve the child’s ability to function daily, positively impacting both the child’s and the family’s quality of life.
A patient with severe HIE (SARNAT type 3) without hypothermia treatment was also associated in our investigation with combined deletion polymorphism (8p11.2 and 14q11.2) and one duplication (10q11.22). The newborn with this genetic condition presented altered MRI scans (with evidence of basal ganglia, central thalamic, and cerebral cortex involvement) and global developmental delay. This polymorphism is a rare genetic disorder that occurs when a small piece of chromosome 8 and 14 is missing, and a small piece of chromosome 10 is gained. These deletions and duplication can occur spontaneously, meaning it is not inherited from the parents, or they can be inherited from a parent who has a balanced translocation involving chromosome 8, 10, and 14. This loss of genetic material can affect the expression of multiple genes, which can lead to a range of physical and developmental features, including intellectual disability, delayed speech and language development, behavioral problems, and distinctive facial features such as a small head circumference, a high forehead, and widely spaced eyes. Additionally, individuals with this condition may have abnormalities of the heart, kidneys, or other organs. Therefore, management and treatment of this condition will depend on the specific features and needs of each affected individual and may involve a multidisciplinary team of healthcare professionals. Treatment may include early intervention and special education programs, physical therapy, occupational therapy, and speech and language therapy, among other interventions.
Besides, one patient had Sotos syndrome, a genetic disorder caused by a mutation in the NSD1 gene that produces a nuclear receptor binding SET domain protein 1. This mutation can occur in different ways, including deletions, duplications, or point mutations. Usually, these mutations are spontaneous, although in some rare cases, they can be inherited. This syndrome is characterized by excessive growth of body tissues, which is reflected in physical features such as a large head, elongated face, and tall stature. In addition, it can affect cognitive and behavioral development, causing intellectual disability and behavioral problems in some people.31 Because it is a rare disorder, treatment must be individualized for each patient, considering their unique needs and challenges. Treatment of Sotos syndrome primarily aims to maximize the patient’s physical, cognitive, and social developmental potential. It may include medical, therapeutic, and educational interventions tailored to the patient’s needs. In addition, periodic monitoring of growth and development, treatment of associated medical conditions, and genetic counseling are some of the medical interventions that may be implemented. Therapeutic interventions, such as speech, occupational, physical, and other behavioral and social interventions, may address cognitive and behavioral challenges.32 In addition, specialized educational treatment may also be necessary to help the patient develop to his or her full potential.
Mutations of uncertain significance (VUS) can present a challenge when diagnosing children with neurodevelopmental disorders. VUS are genetic variants identified in an individual’s DNA whose impact on gene function is unknown. Although VUS is not associated with a disease or disorder, it may increase the risk of developing a condition. In the context of neurodevelopmental disorders, VUS can complicate the diagnostic process because they may be found in children who present with symptoms consistent with a specific disorder, but the significance of the mutation is unclear. In particular, genetic alterations in the ALDH5A1(c.305T>G (p.Val102Gly)) and TH (c.1197+4G>T (Intronic)) genes (both VUS) were found in patients with a SARNAT type 3. These genes provide instructions for producing enzymes found in several metabolic processes. For instance, the ALDH5A1 gene provides instructions for producing the succinic semialdehyde dehydrogenase (SSAD) enzyme, which is involved in the breakdown of a chemical that transmits signals in the brain called gamma-amino butyric acid (GABA). The primary role of GABA is to prevent the brain from overloading with too many signals. Once GABA molecules have been released from nerve cells, they are broken down by SSAD and other enzymes. On the other hand, the tyrosine hydroxylase (TH) gene is essential for making the enzyme necessary to produce dopamine. Dopamine is an important neurotransmitter that plays a role in motor control and movement.33 Mutations in these genes could produce an enzyme with little or no activity34–36 and have heterogeneous neurological consequences ranging from mild to severe.33,37 In our case, the patient with an alteration in SSAD presented language delay, while the patient with an alteration in TH gene presented global developmental delay. Consequently, the carrier state of the patient with the ALDH5A1 gene and the patient with uncertain significance regarding their TH gene cannot explain by their sequelae. Thus, we need additional studies to confirm that sequelae are related to a genetic disorder. This uncertainty can lead to delays in diagnosis and treatment and anxiety for families seeking answers about their child’s condition. However, it is important to note that VUSs are a common finding in genetic testing, and not all VUSs are clinically relevant. In fact, many VUSs are eventually reclassified as benign or pathogenic as more information about their function becomes available.38
Genetic counseling can be helpful for families who receive a VUS result. A genetic counselor can provide information about the likelihood that the VUS is related to the child’s symptoms and help families make informed decisions about follow-up testing and treatment.39 Overall, the impact of mutations of uncertain significance on the diagnosis of children with neurodevelopmental disorders can be significant, as they can complicate the diagnostic process and create uncertainty for families. However, it is important to remember that not all VUS are clinically relevant, and genetic counseling can help families make informed decisions about follow-up testing and treatment.
In addition, support can also be presented through magnetic resonance imaging to find characteristic alterations of the suspected disease. A more accurate way to identify a genetic disorder is by using state-of-the-art sequencing panels, as there is an extensive gene registry for these diseases and a growing understanding of them. However, the time and immediate availability to perform this type of test can be a limiting factor in most cases since a rapid and accurate diagnosis is crucial for survival or to prevent morbidity from increasing in the absence of treatment.40 In summary, a personalized treatment approach involving healthcare professionals, educators, and family members working together is essential to ensure that each patient receives appropriate interventions and support.
The value of differential diagnosis increases as knowledge of the pathologies involved increases and diagnostic tools advance in terms of complexity and time reduction. In addition, children and their families affected by these diseases face difficulties in seeking a correct diagnosis, adequate information, and access to qualified professionals.29 On the other hand, presenting a rare disease entails greater vulnerability in the psychological, social, economic, and cultural spheres. These difficulties could be overcome through appropriate policies or programs. Due to insufficient scientific and medical knowledge, many patients still need to be diagnosed, making it even more challenging to obtain adequate support.
This paper presented the evaluation for patients with HIE. We determine the origin of their clinical phenotype through personalized medicine and correlate the functional and neurodevelopmental outcomes with magnetic resonance imaging (MRI) and genetic findings.
The association obtained between genetic mutations associated with neurotransmitters and the risk of presenting sequelae related to HIE demonstrated the need to determine these and other genetic markers in the development of HIE and to estimate the severity of the developing pathological hypoxic state.
The appearance of a disease, its frequency, and its distribution in different population groups are fundamental pillars for understanding the genetic architecture of human diseases. The genetic and demographic history of rare and harmful genetic variants can be crucial in identifying the risk of suffering from a particular disease. However, inheritance patterns, incomplete penetrance, late appearance, and gene-environment interactions make determining disease risk in populations difficult.
In future works, we plan to extend the study to a large longitudinal cohort to assess properly neurodevelopmental and volumetric findings.
• DG: Methodology, Methods Writing, Original draft preparation.
• NTA: Methodology, Data curation, Investigation, Validation.
• NCR: Methodology, Data curation, Investigation, Validation.
• FRR: Data curation, Writing Original draft preparation.
• CS: Writing Original draft preparation.
• JMEA: Conceptualization, Statistical Methods, Writing Original draft preparation, Reviewing and Editing.
• HFGA: Conceptualization, Statistical Methods, Writing Original draft preparation, Reviewing and Editing.
• GLPH: Conceptualization, Data curation, Methods, Writing Original draft preparation. Writing, Reviewing and Editing.
All procedures performed in the study involving human participants were in accordance with the ethical standards of the Colombian institutional and national research committee and with the 8430-1993 Declaration and its later amendments or comparable ethical standards. The Ethics committee Review Board approved the study at COMFAMILIAR RISARALDA CLINIC (approval no. 00049, date 2019-05-09). The patients’ legal guardians provided written informed consent for publication.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)