Keywords
KRAS mutation, inhibitors, resistance, gastrointestinal cancers, RAS signaling pathway, clinical trails for KRAS
This article is included in the Oncology gateway.
KRAS mutation, inhibitors, resistance, gastrointestinal cancers, RAS signaling pathway, clinical trails for KRAS
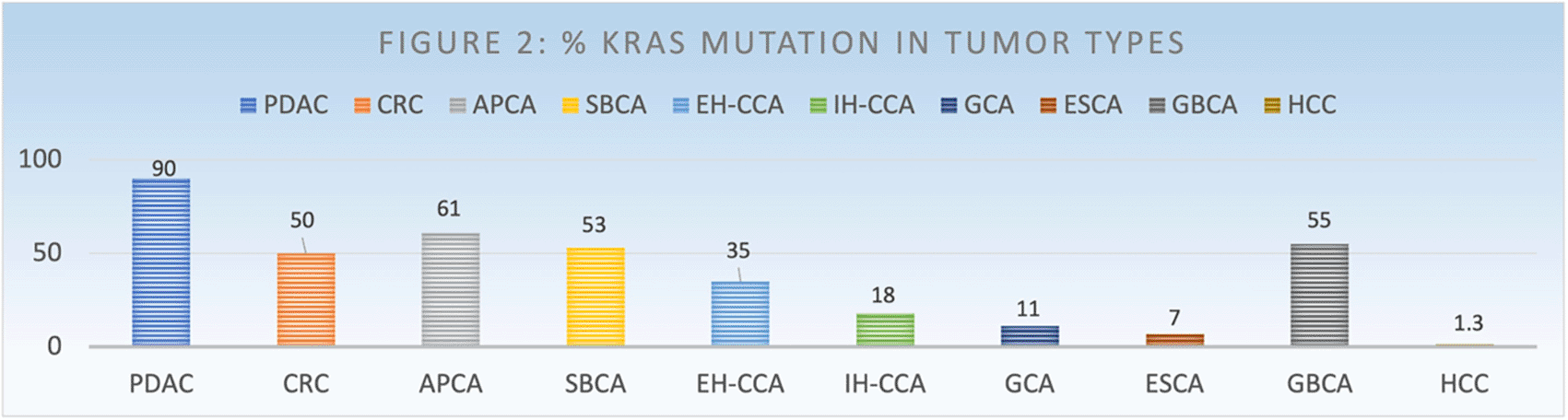
Ras signaling plays an important role in normal physiological cellular proliferation. Activating disarrangement in the RAS pathway can contribute to tumorigenesis and progression from premalignant to invasive malignant pathology (Gurung & Bhattacharjee, 2015). Kirsten rat sarcoma viral (KRAS) oncogene homolog gene mutations are common in gastrointestinal (GI) cancers and lung cancers (Punekar et al., 2022). Colorectal cancer (CRC) is the third most common cancer in the USA. It is projected that CRC will to cause 52,580 deaths and pancreatic cancer will cause 49,830 deaths in the USA in 2023 (Siegel et al., 2022). Over 50% of CRC have mutations in RAS gene and these are associated with worse prognosis. Pancreatic cancer (PDAC) is expected to be the second leading cause of cancer related mortality by 2030. Overall outcome of patients with PDAC has not significantly improved. RAS mutations are present in 80-90% of PDAC and are also associated with worse outcomes. KRAS mutations are also commonly observed in appendiceal cancer (50%), cholangiocarcinoma (23%), gallbladder carcinoma (18%), gastric cancer 11%, esophageal cancer 7%, and HCC (1.3%) (Salem et al., 2021; Prior et al., 2020). As the most common molecular alteration in several GI cancers, successfully targeting RAS mutations could provide significant improvements in GI cancers.
RAS had been considered as a non-druggable therapeutic target. Recent development of novel targeted therapies such as sotorasib and adagrasib against tumors harboring KRAS mutation at the G12C has set the stage for targeting mutated RAS (Hong et al., 2020). Recently reported clinical trials with sotorasib and adagrasib targeting KRASG12C mutation revealed promising anti-tumor activity in previously treated cancers (Hong et al., 2020). The response of different tumor types to KRAS G12C targeted drugs has been variable with best results seen in non-small cell lung cancer (NSCLC) followed by CRC. There have also been variable response rates based on the drug studied. These early results highlight the complexity of targeting KRAS with mutant specific inhibitors for a broad range of malignancies. Several ongoing studies are underway and will provide key information regarding mechanisms of resistance and potential for combination therapy. This review article aims to provide in depth review of preclinical rationale as well as recently completed and ongoing clinical trials of KRAS G12C targeting drugs for RAS-driven GI cancers.
The ras sarcoma (RAS) gene family includes KRAS which is the most commonly mutated gene in this family, other then Harvey rat sarcoma (HRAS) and neuroblastoma rat Sarcoma (NRAS) (Bos, 1989; Lee et al., 2022). HRAS is located at chromosome 11p15.5 and is mostly mutated in cutaneous melanoma (Cox et al., 2014); NRAS is located at chromosome 1p13.1 and mostly mutated in hematopoietic cancers. RAS proto-oncogene is located at chromosome 12p12.1 (Prior et al., 2012). RAS activation starts with surface protein EGFR (epidermal growth receptor) stimulation.
Activation of surface protein growth receptors such as epidermal growth receptor leads to activation of RAS which phosphorylates and activates multiple downstream signaling proteins as discussed below in details that is leading to activation of the Raf- MEK1- ERK, PI3K-AKT- mTOR- NF-κB, and RALGDS-RalA/B pathways (Corral de la Fuente et al., 2022; Dinu et al., 2014). These downstream pathways play critical roles in the tumorigenesis, angiogenesis, differentiation, proliferation, survival, and metastasis of various types of human cancer cells (Figure 1) (Dinu et al., 2014; Jančík et al., 2010; Malumbres & Barbacid, 2003).
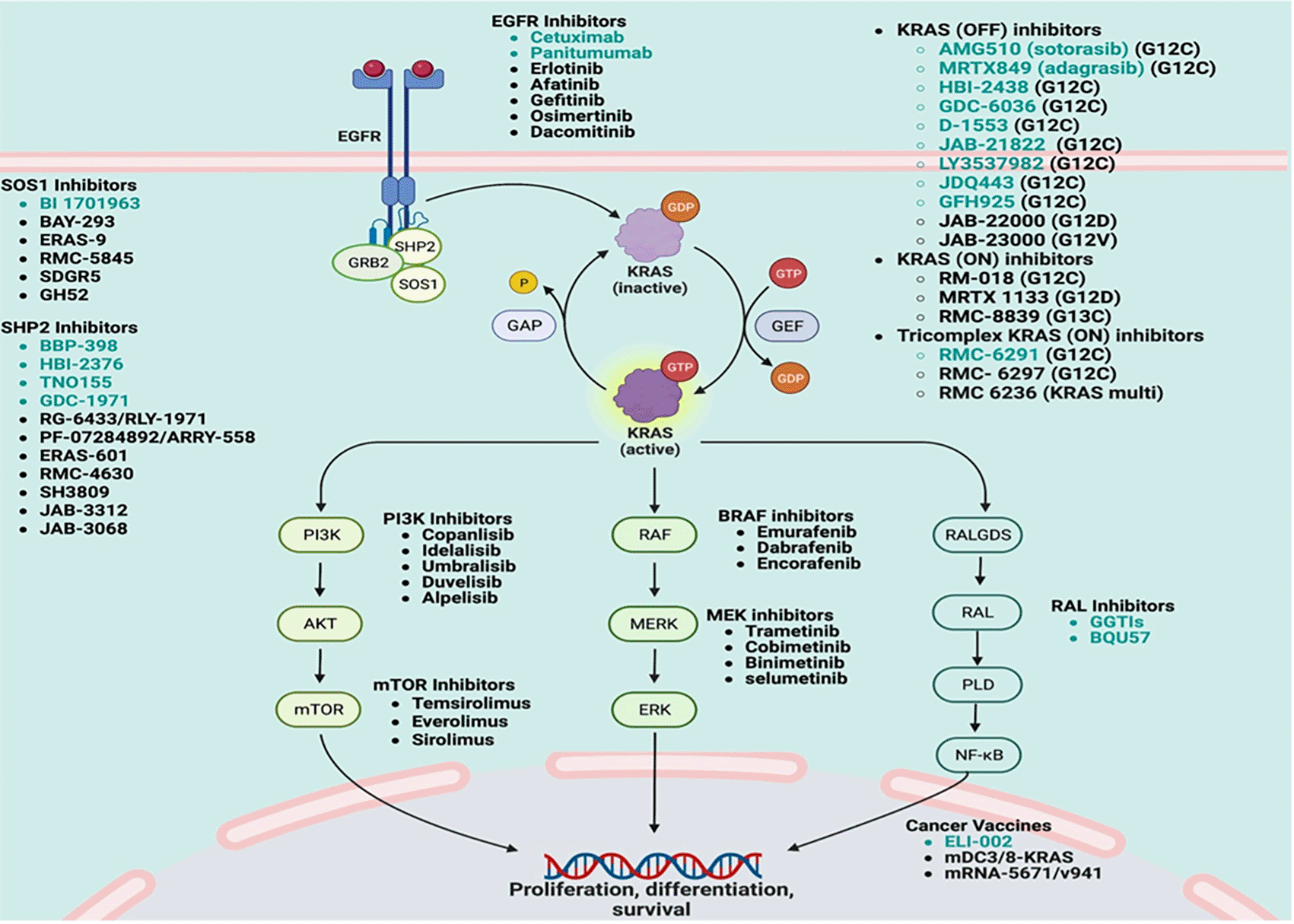
The working mechanism of pathway has been explained in RAS biology section as above. The complexity of the pathway could create several potentials for introducing new agents. Green color-coded agents are being investigated in ongoing clinical trials for GI cancers. This figure is created by using biorender.com.
RAS signaling is complex and has multiple stages (Figure 1). Activation of surface protein EGFR leads activation of RAS by involving multiple proteins at downstream pathways such as SOS1, SHP2, GRB2, GAPs and GEFs etc. EGFR stimulation initially leads dimerization and phosphorylation of EGFR by tyrosine kinase activation which helps GRB2 bind to SOS to stabilize membrane localization via the SH3 domain (Parikh et al., 2022; Aronheim et al., 1994). The interaction between RAS, son of seven less 1 (SOS1) and SHP2 protein-tyrosine phosphatase interaction are an important mediator of cellular signaling through the RAS/MAP kinase (Hofmann et al., 2021). SHP2 activation by EGFR-SOS1 communication stimulates GEFs (guanine nucleotide-exchange factors) which converts guanosine diphosphate (GDP) to guanosine triphosphate (GTP) which primes activation of RAS by conformational modification in the switch and endorses downstream signaling (Boriack-Sjodin et al., 1998; Chen et al., 2016; Parikh et al., 2022; Wolfman & Macara, 1990). GTP hydrolysis to GDP is facilitated by GTPase-activating proteins (GAPs), which triggers the deactivation of RAS (Cherfils & Zeghouf, 2013). That is why inhibition or depletion of SOS1 and SHP2 have impact on regression of tumor growth (Hofmann et al., 2021; Jeng et al., 2012). In addition, oncogenic RAS mutations causing resistance GTPase and GEFs leads to constant KRAS activation which leads to tumorigenesis (Boriack-Sjodin et al., 1998; Parikh et al., 2022).
There are multiple different types of KRAS inhibitors, including KRAS (ON), KRAS (OFF) and pan-KRAS inhibitors to overcome mutations in KRAS (Figure 1). KRAS (OFF) inhibitors target the inactive form of KRAS by locking it in its inactive state, preventing the transmission of downstream signals (Boriack-Sjodin et al., 1998; Oyedele et al., 2022; Parikh et al., 2022). KRAS (ON) inhibitors work on active form of GTP-bound KRAS by blocking passing the downstream signals which can be more effective and faster than “OFF” inhibitors sometimes. Pan-KRAS (tricomplex KRAS) inhibitors work on both active and inactive from of KRAS by using a novel tri-complex formation (Parikh et al., 2022).
The tumor microenvironment (TME) hosts a dynamic field of interaction between immune, stromal, and tumor cells. The interactions between the tumor cells and the TME plays a role in tumor proliferation, metastasis, immune surveillance, and resistance to therapy. KRAS plays a crucial role in TME through activation several transcriptional regulators such as NK-kB which stimulates pro-inflammatory (ICAM-1, TNF-α, and IL-18) and anti-inflammatory (IL-10, IL6, TGF-β, and GM-CSF) cytokine production (Pereira et al., 2022). KRAS mutated pancreatic cancers have high expression of intracellular adhesion molecules (ICAM) which leak into TME as soluble ICAMs. ICAMs contribute to degradation of extracellular matrix (ECM) further leading pancreas cell to differentiate and lead to metaplasia (Pereira et al., 2022). In addition, several studies have shown that mutant KRAS plays a role in anti-inflammatory and immunosuppressive processes by releasing some cytokines. The increased expression of anti-inflammatory cytokines drives the development of the commonly observed PDAC immune-inhibitory microenvironment characterized by inhibitory tumor associated macrophages (TAM), myeloid cells, T-regs, and exhausted lymphocytes. Specifically, KRASG12D mutations increase anti-inflammatory cytokine that suppresses cytotoxic CD8+ T cell-mediated tumor and increases immunosuppressive T-regs. KRAS mutated cell decreases MHC 1 expression and upregulates programmed cell death 1 (PD-1), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and PDL 1 expression in TME which lead to weak recognition of cytotoxic T cell to kill tumor cells (Cheng et al., 2019; Hamarsheh et al., 2020; Pereira et al., 2022). KRAS mutant cancer cells also have low tumor mutational burden (TMB) which can limit potential for immunotherapy (Liu et al., 2020). In addition, IL6 is produced in KRAS mutated pancreas cells and triggers immunosuppressive condition which creates an environment to initiate the pancreas cancer development (Ancrile et al., 2007). In lung adenocarcinoma PD-L1 is expression is higher with the TP53/KRAS co-mutation which can potentially increase efficacy of immunotherapy (Dong et al., 2017). Moreover, KRAS inhibitors also propagate TME by increasing T cells, macrophages, and dendritic cells (Canon et al., 2019).
Different methods are employed to detect KRAS mutations such as RT-PCR, NGS, Sanger sequencing, pyrosequencing, BEAMing technique, and dideoxy nucleotide sequencing which can be done either primary sites or metastatic sites with 93 % concordance rate of mutations (Wong et al., 2014).
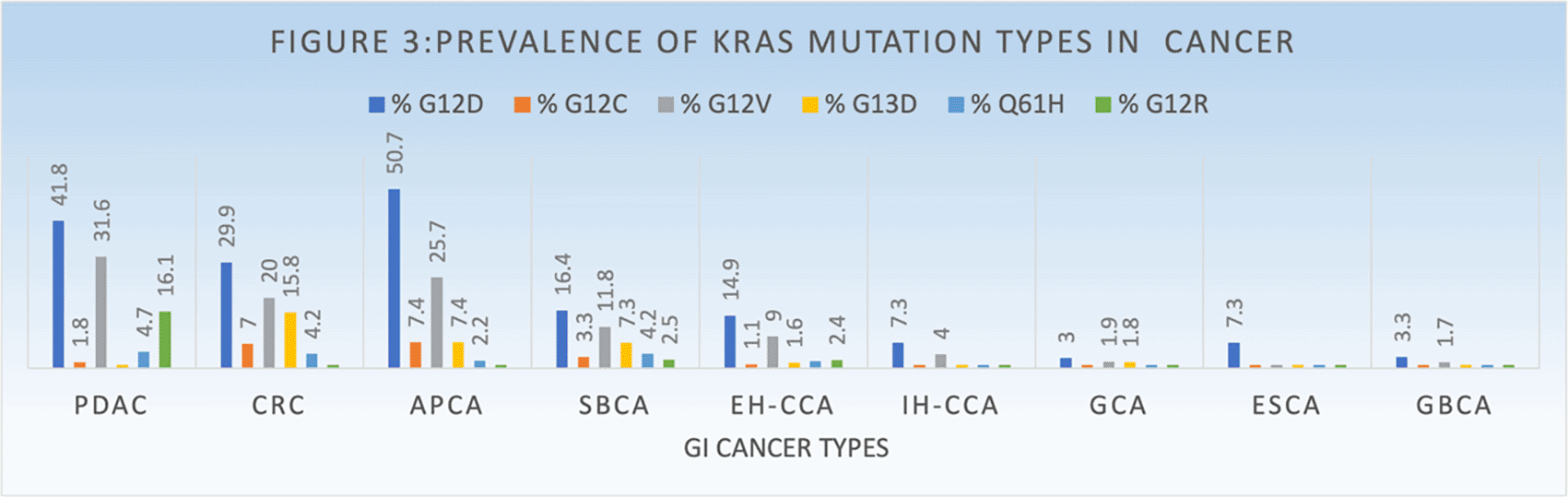
KRAS mutations mostly occur in codon 12 (77%), such as subtypes of G12D, G12V and G12C, and rarely occur in codon 13, such as G12D subtypes. Each subtype is associated with different pathway activations, for example G12D and G12V activate Ral A/B pathway in smokers, G12D activates PI3K/AAKT and MEK pathway in nonsmokers (Adderley et al., 2019; Friedlaender et al., 2020). Some studies showed that KRAS can be co-mutated with genes (TP53-39%, STK11-20%, and KEAP1-13%) during oncogenesis, which can play a role in aggressiveness of tumor and poorer treatment response (Corral de la Fuente et al., 2022). KRAS mutational variants differ based on the primary site of GI cancers such as G12D (35.4%), G12V (23.5%), G12R (8.7%), G13D (8.0%), Q61H (4.6%), and G12C (4.3%) (Figures 2 and 3). Colorectal cancers have G12D (15–29.9%), G12V (10–20.0%), G13D (8.1–15.8%), G12C (3.6–7.0%), G12A (4.9%), G12R (<1%) and Q61H (4.2%). In PDAC, prevalence of KRAS mutations variants is G12D (39.5–41.8%), G12V (28.6–31.6%), G12R (14.6–16.1%), Q61H (4.7%), G13D (<1%) and G12C (1.5–1.8%). KRAS mutation prevalence in appendiceal cancer is as follows: G12D (≈30–50.7%), G12V (16.3–25.7%), G12C (3.1–7.4%), G13D (5–7.4%), G12S (2.9%), and Q61H (2.2%) (Hong et al., 2020; Johnson et al., 2020; Lee et al., 2022; Salem et al., 2021; Zhou et al., 2022). In addition, a very comprehensive study from the Precision Oncology group regarding KRAS mutation prevalence has reported that small bowel adenocarcinoma has G12D (16.4%), G12V (11.8%), G13D (7.3%), G12C (3.3%), G13D (1.6%), G12R (2.5%). Extra-hepatic cholangiocarcinoma has G12D (14.9%), G12V (9%), G13D (1.6%), G12C (1.1%), G12R (2.4%) while intra-hepatic cholangiocarcinoma has G12D (7.3%), G12V (4%), G13D (<1%), G12C (<1%), G12R (<1%) and gallbladder adenocarcinoma has G12D (3.3%), G12V (1.7%), G13D (1%), G12C (<1%), G12R (<1%). Esophageal adenocarcinoma has G12D (7.3%), G12V (<1%), G13D (1.1%), G12C (<1%), G12R (<1%) and gastric adenocarcinoma has G12D (3%), G12V (1.9%), G13D (1.8%), G12C (<1%), G12R (<1%) (Lee et al., 2022).
Several preclinical studies have revealed promising results with KRAS inhibitors in GI cancers, mainly targeting KRASG12D and KRASG12C. A preclinical study examining KRASG12D inhibitor (MRTX1133 and MRTX849) in a KRASG12D mutant xenograft mouse tumor model revealed promising anti-cancer activity in PDAC (Hallin et al., 2020; Wang, X. et al., 2021) and more recently MRTX-1133 demonstrated promising efficacy in immune competent mouse models of PDAC (Kemp et al., 2023). Both MRTX-1133 and AMG-510 demonstrate potent modulation of anti-tumor immunity in immune competent mice bearing KRASG12C CT26 and KRASG12D 2838c3/6419c5 colorectal and PDAC tumors respectively, inducing potent tumor regression that was dependent on accumulation of cytotoxic tumor infiltrating CD8+ and CD4+ T cells. Furthermore, in both models there was evidence of a shift in polarization of macrophage to a cytotoxic M1 phenotype and increases antigen presentation through dendritic cells (Canon et al., 2019). Previous studies involving the use of KRAS deficient or sufficient KPC cell lines in immune competent mice support a critical role for oncogenic KRASG12D signaling in programing of an immune suppressive microenvironment critical for pancreatic tumor progression (Ischenko et al., 2021). The immunomodulatory role of KRAS inhibitor was further studied in mice with CT-26 KRASG12C mutated tumors in which combination of AMG-510 with PD-1 inhibitor boosted anti-tumor T cell activity and significantly decreased tumor progression indicating synergy with immune checkpoint blockade. In the CT26 model, AMG-510 also stimulated expression of chemokines (Cxcl10 and Cxcl11), which are crucial attractants of tumor-suppressive immune cells (Canon et al., 2019; Wang, & Fakih, 2021). Importantly the anti-tumor immune activity of adagrasib and sotrasib and synergy with PD-1 ICB were dependent upon the presence of intrinsic interferon signaling in tumor cells in preclinical models of NSCLC (Mugarza et al., 2022) highlighted a critical role for the immunogenicity of tumor of the tumor immune microenvironment as a critical factor in synergy between immune checkpoint therapy and mutation-specific, oncogenic KRAS inhibitors.
An additional novel technique is development of iExosomes which harbor siRNA or shRNA exclusive to target oncogenic KRASG12D in genetically engineered KRASG12D mutant mouse models of pancreatic cancer (Kamerkar et al., 2017). Those iExosomes inhibit generation of oncogenic KRAS protein through an siRNA knockdown, resulting in inhibition of KrasG12D-signaling in KRASG12D mutated tumors reducing metastases and improving overall survival (Kamerkar et al., 2017). This preclinical research led to a clinical trial of iExosomes for treating metastatic pancreas cancer with KRASG12D mutation (NCT03608631). Adoptive cell transfer of TILs or PBL, directed towards the KRAS pathway, has been employed in preclinical models as another form of immune-based therapy targeting the KRAS pathway (Chatani & Yang, 2020). Furthermore, bispecific antibodies that have been genetically engineered to bind to HLA-restricted epitopes and TCR have been suggested for GI cancers, but they have not yet been tested in clinical trials for colon cancer. In preclinical studies, bispecific antibodies that recognized linked HLA alleles were found to be effective in killing human cancer cell lines with KRAS mutations (Douglass et al., 2021).
Prior studies aimed at target KRAS mutations failed to achieve meaningful results in GI cancers due to early terminations secondary to no objective responses or unexpected toxicity and were withdrawn by investigators (Table 1). Fortunately, preliminary data in early phase clinical trials showed some promising results in GI cancers by targeting KRAS mutation in KRYSTAL 1 and CodeBreak100 clinical trials as outline in Table 2.
| Trials ID | Phase | Eligibility criteria | Year | Enrolled patients | Treatment arm | Control arm | Outcome | Median follow-up (year) |
|---|---|---|---|---|---|---|---|---|
| NCT00326495 | II | KRAS mutation EGFR expressed Failed first line mCRC | 2014 | 51 | cetuximab and sorafenib | NA-Single arm | Terminated-No objective responses were observed | 1.84 months |
| NCT01109615 | II | KRAS mutation Failed first line mCRC | 2012 | 40 | Pemetrexed and Gemcitabine | NA-Single arm | Terminated- Lacking effect of treatment | NR |
| NCT04627142 | I | KRAS mutation Failed first line mCRC | 2022 | 15 | BI 1701963 (Turn off KRAS) and Irinotecan | NA | Terminated-Sponsor decision | NR |
| NCT01365910 | II | KRAS mutation Failed first line mCRC | 2013 | 30 | Linifanib (MRTKI) | NA | Terminated-Interim analysis determined the study did not meet criteria to proceed | 16.4 weeks |
| NCT04165031 | 1/II | KRAS mutation Failed first line mCRC | 2020 | 5 | LY3499446 (KRAS G12C inhibitor) | NA | The study was terminated due to an unexpected toxicity finding. | NR |
| NCT01646554 | II/III | KRAS mutation Failed first line mCRC | 2016 | 0 | FOLFOX + Aflibercept | FOLFOX | Withdrawn | NR |
| NCT01562899 | Ib/II | KRAS mutation Failed first line mCRC PDAC | 2015 | 77 | MEK162 (Binimetinib) + AMG 479 (Ganitumab) | NA | Terminated (Study was withdrawn due to scientific and business considerations.) | NR |
| NCT02448810 | IIa | KRAS mutation Failed first line mCRC | 2017 | 115 | Anti-MIF/BAX69 (imalumab) + infusional 5-FU/LV | NA | Terminated (Based on overall benefit-risk assessment.) | 24.9 weeks |
| Trials ID/Name | Eligibility criteria | Treatment intervention, single arm | Median follow-up | mPFS/DFS/OS | ORR/DCR (%) | Grade 3/4 TRAEs (%) |
|---|---|---|---|---|---|---|
| NCT03600883 (CodeBreak 100) Phase I/II | KRAS G12C Mutation CRC arm | Sotorasib (n=42) | 12.8 months | 4 months (mPFS) | 7.1/73.8 | 12% pts |
| KRAS G12C Mutation PDAC arm | Sotorasib (n=38) | 16.8 months | 4 (mPFS)/ 7 (mOS) months | 21.1/84.2 | 15% pts | |
| NCT04793958 (KRYSTAL-1) Phase I/II | KRAS G12C Mutation mCRC arm | Adagrasib (n=45) | 8.9 months | 5.6 months (PFS) | 22/87 | 30% pts |
| Adagrasib + Cetuximab (n=36) | 7 months | NR | 43/100 | 16% pts | ||
| NCT01188785 phase I/IIa | KRAS G12C Mutation LAPC | RNAi Therapy (siG12D LODER) + Gemcitabine + mFOLFIRINOX (n=15) | 8 weeks | 15 months (mOS) | NR | 11 % pts |
KRYSTAL 1 (NCT03785249) is a phase I/II clinical trial designed to evaluate safety and efficacy of highly selective KRAS inhibitor, adagrasib (MRTX849) in non-small-cell lung cancer, colorectal cancer, and other solid tumors. The results of the trial from CRC arm revealed that adagrasib (MRTX849) achieved 87% DCR (disease control rate) and 22% ORR (overall response rate) as monotherapy while combination arm showed cetuximab with adagrasib (MRTX849) achieved 100% disease control rate (DCR) and 43% objective response rate (ORR) in heavily pretreated KRASG12C mutated colorectal cancers (Weiss et al., 2021). The CodeBreaK100 (NCT03600883) trial with sotorasib as monotherapy enrolled more KRASG12C-mutated locally advanced and mCRC patients (n=42), 98% of those enrolled CRC patients had failed first line treatments. Median follow-up in this study subgroup analysis was 12.8 months. A DCR of 74 % and ORR of 7.1% was reported in the CRC arm. Progression free survival (PFS) was 4 months (Hong et al., 2020). The phase II single-arm CodeBreaK100 clinical trial investigated the safety and activity of sotorasib monotherapy in 62 KRASG12C-mutated advanced CRC patients who failed fluoropyrimidine, oxaliplatin, and irinotecan-based first line therapy. The treatment was well tolerated, and 9.7% ORR was reported (Fakih, M. G. et al., 2022).
Pancreatic cancer is one the most challenging GI cancers and had poor prognosis that has good potential regarding targeting KRAS due to prevalence of the KRAS mutations. In a recent 2022 ASCO Plenary Series, KRASG12C inhibitor (sotorasib) showed meaningful results in patients with pretreated KRASG12C-mutated pancreatic cancer from combined phase I/II CodeBreaK100 (NCT03600883) clinical trial. Sotorasib as monotherapy had ORR of 21.1% and DCR was 84.2% with acceptable safety profile (Strickler et al., 2022). Moreover, other strategy to target KRAS mutation is using RNAis. RNAi therapy targeting KRASG12D with concurrent chemotherapy was shown to be efficacious with mOS of 15 months in locally advanced pancreas cancer (LAPC) in open label phase I/IIa clinical trial (NCT01188785). This study was designed to use a biodegradable implant called siG12D-LODER™ that was implanted into LAPC and released a siRNA drug against KRAS(G12D) (Golan et al., 2015).
After FDA approval of KRASG12C inhibitors for NSCLC and promising results from KRYSTAL 1 and CodeBreak 100 for GI cancers, several other clinical trials were launched. For example, open-label randomized phase III KRYSTAL-10 (NCT04793958) trial compares adagrasib plus cetuximab versus chemotherapy in previously treated mCRC and a multicenter open-label randomized phase II CodeBreak300 (NCT05198934) trial compares sotorasib and panitumumab versus investigator’s choice of therapy (trifluridine and tipiracil or regorafenib) mCRC. Moreover, multiple clinical trials are underway combining KRAS inhibitors with SHP2 (BBP-398, HBI-2376, TNO155 and GDC-1971), SOS1 (BI 1701963), EGFR inhibitors (cetuximab and panitumumab) and PD-1/PDL-1 inhibitors as outlined in Table 3. Increased expression of PD-L1, PD-1 and CTLA in KRAS mutated GI cancer cells and potentially immunomodulatory effects of KRAS inhibitors provides rationale to combine KRAS inhibitors and immunotherapies such as immune checkpoint inhibitors, adoptive T cell transfer and cancer vaccines. Table 3 includes some of ongoing clinical studies to overcome KRAS resistance with combination strategy. For example, AMG 404 (anti PD-1) plus sotorasib versus sotorasib alone in advanced CRC and PDAC (NCT04185883), adagrasib plus pembrolizumab or cetuximab or afatinib versus adagrasib alone in advanced CRC and PDAC (NCT03785249), LY3537982 plus EGFR inhibitor or PDL-1 inhibitor or SHP inhibitor versus pembrolizumab alone in advanced KRASG12C-mutant solid tumors (NCT04956640) and JDQ443 plus tislelizumab (anti PD-1) or SHP inhibitor versus JDQ443 in advanced CRC (NCT04699188) are examining this potential. NCT03745326 and NCT03190941 examine anti-KRASG12D and G12V mTCR PBL (Peripheral Blood Lymphocytes) in KRAS mutated GI cancers. Other two phase I clinical trials are designed to use mRNA plus pembrolizumab and DC vaccine in pancreatic cancer (NCT03948763, NCT03592888).
| Trials name and ID | Cancer types | Estimated Enrollment | Targeting KRAS pathway | Control arm | Phase | Start and completion dates | Primary measures |
|---|---|---|---|---|---|---|---|
| NCT04185883 (CodeBreak 101) | CRC PDAC Solid Ca | 1054 | Sotorasib + | Sotorasib alone | I and II | 12/2019-10/2026 | ORR, DLTs, AEs |
| NCT05198934 (CodeBreak 300) | CRC | 153 | Sotorasib and Panitumumab | Investigator's Choice (Trifluridine and Tipiracil, or Regorafenib) | III | 4/2022-3/2024 | PFS |
| NCT05251038 | PDAC | 59 | Sotorasib + Lip. Irinotecan + 5 FU+ Leucovorin OR Sotorasib + Gemcitabine + Nab-paclitaxel | NA | Ib and II | 11/2022-2/2025 | ORR |
| NCT05480865 (Argonaut) | Solid CA | 85 | Sotorasib + SHP2 Inhibitor (BBP-398) | NA | I | 7/2022-6/2025 | MTD, DLTs, AEs |
| NCT05485974 | CRC PDAC | 44 | HBI-2438 (KRAS G12C inhibitor) | NA | I | 8/2022-8/2025 | MTD, DLTs, AEs |
| NCT05163028 | CRC PDAC | 42 | HBI-2376 (SHP2 Inhibitor) | NA | I/ | 12/2021-12/2024 | MTD, DLTs, AEs |
| NCT03190941 | CRC PDAC GICA GCA RECA | 110 | Anti-KRAS G12V mTCR PBL | NA | I and II | 9/2017-6/2029 | DLTs, PR, CR |
| NCT03745326 | CRC PDAC GCA RCA | 70 | Anti-KRAS G12D mTCR PBL | NA | I and II | 5/2019-12/2028 | DLTs, PR, CR |
| NCT05462717 | CRC PDAC | 117 | RMC-6291(KRAS-G12C(ON) Inhibitor) | NA | 1b/ | 9/2022-12/2025 | AEs, DLTs |
| NCT03785249 (KRYSTAL-1) | CRC PDAC | 740 | Adagrasib + | Adagrasib alone | Ib and II/ | 1/2019-9/2023 | DLTS, ORR, AEs |
| NCT04975256 (KRYSTAL 14) | CRC | 100 | Adagrasib (MRTX849) +BI 1701963 (SOS1 inhibitor) | NA | 1b | 12/2021-11/2023 | DLTs, AEs |
| NCT05379985 | CRC PDAC | 141 | RMC-6236 (RASMUTLI (ON) inhibitor) | NA | I | 5/2022-12/2025 | AEs, DLTs |
| NCT04793958 (KRYSTAL-10) | CRC | 410 | Adagrasib +cetuximab | mFOLFOX or FOLFORI | III | 3/2021-92022 | OS, PFS |
| NCT04330664 (KRYSTAL 2) | Solid CA | 86 | Adagrasib and TNO155 (SHP2 Inhibitor) | NA | I and II | 4/2022-10/2022 | DLTs, Pharmacokinetic and AEs |
| NCT05178888 (KRYSTAL-16) | Solid CA | 50 | Adagrasib+ Palbociclib | NA | Ib | 2/2022-3/2024 | DLTs, ORR, Pharmacokinetic and AEs |
| NCT04853017 (AMPLIFY-201) | PDAC | 18 | ELI-002 immunotherapy (a lipid-conjugated immune-stimulatory oligonucleotide [Amph-CpG-7909] | NA | I | 10/2022-12/2024 | MTD, AEs |
| NCT03948763 | CRC PDAC | 70 | mRNA-5671/V941+ Pembrolizumab | mRNA-5671/V941 | I | 6/2019-8/2022 | DLTs, AEs |
| NCT03592888 | PDAC | 12 | mDC3/8-KRAS Vaccine | NA | I | 11/2018-9/2022 | Safety and side effects of vaccine |
| NCT04449874 | CRC Solid Ca | 498 | GDC-6036 (KRAS G12C inhibitor) + | GDC-6036 (KRAS G12C inhibitor) | I | 7/2020-8/2023 | DLTs, AEs |
| NCT04585035 | Solid CA | 200 | D-1553 (KRASG12C inhibitor) + others | D-1553 alone | I and II | 10/2020-2/2023 | DLTs, AEs and Pharmacokinetics |
| NCT05002270 | CRC Solid Ca | 100 | JAB-21822 (KRAS G12C inhibitor) + Cetuximab | JAB-21822 | I and II | 9/2021-7/2023 | DLTs, ORR, AEs, DOR |
| NCT05009329 | Solid CA | 144 | JAB-21822 (KRAS G12C inhibitors) | NA | I and II | 7/2021-122025 | DLTs, AEs, Pharmacokinetics |
| NCT04956640 | Solid CA | 360 | LY3537982 (KRAS-G12C inhibitor) + | LY3537982 | I | 7/2021-11/2023 | DLTs |
| NCT04699188 | CRC Solid CA | 425 | JDQ443 (KRAS G12C inhibitor) + | JDQ443 | I and II | 2/2021-8/2024 | DLTs, ORR, AEs |
| NCT05005234 | Solid CA | 128 | GFH925 (KRAS G12C inhibitor) | NA | I and II | 9/2021-4/2024 | DLTs |
Development of KRAS resistance is a major challenge for the success of KRAS inhibitors and it is classified into four groups: On-target (1); secondary mutations of binding pocket (R68S, H95D/Q/R or Y96C) genes which prevent medications to bind and activating mutations of KRAS (G12D, G12V and G13D), Q61H) and increased KRAS amplification which overcome inhibition effect. Off-target (2); MET amplification, activating mutations in NRAS, BRAF, MAP 2K1 and RET; oncogenic fusions involving ALK, RET, BRAF, RAF1, and FGFR3 which can lead hyperactivation of with KRAS-MAPK signaling pathway from alternative path. Intrinsic resistance mechanisms (3) can arise from RAS-independent tumors and clonal selection. Additionally, histologic transformation (4) to squamous cell carcinoma in non-small cell lung cancer (NSCLC) is another noteworthy phenomenon (Awad et al., 2021; Corral de la Fuente et al., 2022; Tanaka et al., 2021). MET amplification and loss-of-function mutations in NF1 and PTEN are other type of resistance mechanism in CRC versus other solid cancers (Awad et al., 2021). For example, 38 solid cancer patients with KRASG12C mutation are progressed on adagrasib or sotorasib monotherapy due to later secondary mutations of binding pocket (R68S and Y96C) genes (Awad et al., 2021; Hong et al., 2020). Other studies revealed KRASG12C inhibitors can develop resistance due to binding some of the compounds to cysteine 12 within the switch II pocket of the GDP-bound form of the KRASG12C protein (Awad et al., 2021; Lito et al., 2016). Blocking KRAS pathway can stimulate overexpression of immune responses that can play a role in infiltration of suppressive immune cell in tumor microenvironment (TM). Additionally, combining sotorasib with anti-PD-1 therapy enhanced CD8+ T cells, macrophages, and dendritic cells activity in tumor microenvironment of animal mouse models (Akhave et al., 2021).
There are multiple strategies proposed so far to overcome the resistance mechanism such as using tricomplex KRAS inhibitors, combination with ICI or/and targeted gene mutations. For example, RM-018 is a “tricomplex” KRAS inhibitor that is developed to overcome resistance of KRASG12C/Y96D conferred resistance to multiple KRASG12C inhibitors. RM-018 binds specifically to the GTP-bound, active “RAS (ON)” state of KRASG12C instead of binding inactive state (Tanaka et al., 2021).
One of resistance mechanisms to KRAS inhibitors is upregulation of different receptor tyrosine kinases (RTKs) other than EGFR and their ligands, which reactivates the inhibited pathway (Fedele et al., 2018; Ryan et al., 2020). SHP2 and KRAS (OFF) inhibitors work synergistically to overcome the resistance (Corral de la Fuente et al., 2022; Dunnett-Kane et al., 2021). Combination of KRAS inhibitors with SHP2 and SOS1 inhibitors and immunotherapy can overcome those resistance, which is studied in clinical trials of NCT04330664, NCT04185883, and NCT03785249. For example, ongoing CodeBreak101 (NCT04185883) clinical trial will compare AMG 510 as monotherapy to combined with the EGFR/HER2 TKI-afatinib, or with the anti-EGFR (panitumumab) with or without chemotherapy (Fakih et al., 2020). KRYSTAL-10 (NCT04793958) will be comparing combination of adagrasib and cetuximab with or without chemotherapy.
The role of SHP2 and SOS1 have been investigated in multiple studies. SHP-2 suppress the function of KRAS inhibitors by dephosphorylations and are augmenting the activity of other pathways such as PI3K and MAPK/ERK to overcome KRAS inhibitors (Agazie & Hayman, 2003; Zhang et al., 2004). SOS proteins play a role in activation of RAS-GTP complex to overcome inhibition of KRAS (Rojas et al., 2011). Some of SOS1 inhibitors such as TNO155, BBP-398, and GDC-1971 are currently under investigation in combination setting in clinical trials respectively NCT04330664, NCT05480865, and NCT04449874. Reactivation of the mTOR pathway was shown play a role in resistance of KRAS inhibitors (Brown et al., 2020), which can be another alternative strategy to overcome resistance to KRAS inhibitors by combining mTOR inhibitors. The mTOR inhibitor, everolimus is under investigation with sotorasib combination on CodeBreak 101 clinical trial (NCT04185883). The ongoing clinical trials that are designed to overcome KRAS resistance are listed in Table 3.
The success of targeting KRAS G12C ushered an era of RAS targeted therapies. There are several promising agents currently in development that will engage the more prevalent RAS mutations in GI malignancies. Preclinical and clinical data suggests that RAS mutations are driver mutations which contribute to the biology of tumor microenvironment. Targeting RAS may have impact on tumor progression as well as change the immune and stromal elements in the TME. As these agents are being developed it will be crucial to understand the biology of primary and acquired resistance, which will in turn help develop rational combination therapy strategies.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)