Keywords
Advanced Cervical Cancer, Treatment, Hazard Risk, Relative Risk, Absolute Risk, Number-To-Treat.
This article is included in the Oncology gateway.
Advanced Cervical Cancer, Treatment, Hazard Risk, Relative Risk, Absolute Risk, Number-To-Treat.
Worldwide, cervical cancer is the fourth in incidence and mortality in women. Estimates indicate that in the year 2020, there were 604,000 and 342,000 cases and deaths, respectively. The epidemiology of this malignancy is linked to the economic development of countries and regions. The rates in developed and developing countries vary from 18.8 vs. 11.3 per 100,000 and 12.4 vs. 5.2 per 100,000 for incidence and mortality, respectively. These differences occur even within high-income countries such as the USA, where the death rate from cervical cancer is twice as high among women living in high than those in low-poverty areas. These epidemiological differences are mainly due to deficient cervical cancer screening programs. However, cancer treatment affordability is also an issue.1
Systemic chemotherapy remains the cornerstone of treatment for patients with advanced (metastatic, recurrent, or progressive disease) diseases who are not candidates for salvage surgery or radiation. Advanced cervical cancer is a tumor in which the benefit of systemic chemotherapy over best palliative care was never established from randomized studies. A seminal study by Thigpen in 1981 administering 50 mg/m2 of cisplatin every three weeks reported a median overall survival (mOS) of six and nine months for the responders and not responders, respectively. The median OS for the whole patient population was not reported.2 Bonomi et al. in 1985 compared three cisplatin schedules: 50 mg/m2 (d1), 100 mg/m2 (d1), and 20 mg/m2 d1-5 (100 mg/m2 total). There were significant differences in response rates; however, the median OS of 7.1, 7.0, and 6.1 months were similar.3 On this basis, cisplatin 50 mg/m2 every 21 days was accepted as the standard of care against further studies that should be compared.
In 2005 cisplatin-topotecan increased mOS over cisplatin alone from 6.5 to 9.4 months4 and was established as the standard of care. Afterward, in 2009, the cisplatin-paclitaxel doublet showed a mOS of 12.8 compared with three other doublets (cisplatin-vinorelbine, 10.8 months, cisplatin-topotecan 10.2 months, cisplatin-gemcitabine 10.2 months5 and was recommended as the standard of care. In 2014, the new suggested standard was bevacizumab added to chemotherapy, either cisplatin-paclitaxel or paclitaxel-topotecan, proving an increase in mOS from 13.5 to 17 months.6 The most recent improvement was the KEYNOTE-826 trial, when pembrolizumab proved superior to cisplatin-paclitaxel with or without bevacizumab (mOS 24 vs. 16.4 months).7
Globally, a central problem with new cancer treatments is their price. The high cost causes a significant disparity in access to treatment. For most patients worldwide, some treatment options may be insufficient or nonexistent. For those who can afford treatment, the cost of treatment can cause financial toxicity. A study has shown that resulting financial toxicity increases the Risk of death from the cancer the patient is being treated.8 The monthly cost of new chemotherapy agents is 12,000 USD https://www.cancer.gov/news-events/cancer-currents-blog/2018/presidents-cancer-panel-drug-prices. To put this number in perspective, according to the website world data, the median monthly income for 69 countries is 881 USD (maximum of (15,507 in Luxembourg to 42 in Afghanistan) https://www.worlddata.info/average-income.php). It can be realized that 12,000 USD monthly is unattainable for individuals and many health systems. To better understand the high price of drugs phenomenon, we briefly overview the situation in the recent past and how it has evolved regarding clinical cancer research and cancer treatment.
In the history of the development of pharmacological treatments for cancer, there was a point where this development ceased to be a genuinely humanist scientific activity heavily financed by the Governments to another where science has become the instrument for pharmaceutical and biotechnological corporations for lucrative activity. Since the early years after World War II, when the first chemotherapeutic agents were discovered, until the 1990s, advances in cancer research enabled and encouraged the development of virtually all the chemotherapeutics we use today. Pharmaceutical companies were limited to their well-established and indispensable role of completing preclinical and clinical development and their subsequent registration and commercialization at prices that, while relatively expensive compared to drugs from other areas of medicine, were relatively affordable. The prominent cancer cooperative groups in the western world (mainly the USA, Western Europe, and Japan) were sponsored by public funds and designed and carried out the most critical clinical trials. At that time, pharmaceutical companies generally limited themselves to providing the study drug without significant influence on the design, study conduct, or publication of results.
When the studies were positive, and once the products were commercialized, further clinical trials on the drug aimed to optimize the treatments, for example, studying new combinations, duration, and intensity of the treatments, concurrent vs. sequential schedules, and, of course, head-to-head comparisons to establish their efficacy and toxicity. In other words, medical oncologists interested in optimizing the treatments originated the studies with no more influence from the industry providing the research product at most.
On the other hand, the primary role of Institutional Ethic Committees was to comply with the ethical requirements of any human study in the shortest possible time and with the fewest possible bureaucratic requirements. They knew that promoting and accelerating clinical research was an ethical imperative for the medical researchers and the ethics committees. Many of the oncology clinical trials were originated by the investigator or groups of investigators, and they could carry it out due to minimal necessary regulation. Of course, at that time, there was no need for Contract Research Organizations (CROs); needless to say, clinical trials were less expensive than now.
During that time, the vast majority of the chemotherapy treatment protocols we have today were established, which allowed many tumors to be cured or at least increased survival. Starting in the 1990s, the free market in health began to interfere with each one of the processes involved in the pharmacological therapy of cancer. One of the first consequences was the reduction of public funds for the research and development of cancer therapies, clearing the way for increased private capital to finance the research and development of cancer treatments. This situation was accompanied by a sharp increase in drug prices.9 This new “form” of clinical research has become a genuine industry where what matters least is that they are affordable treatments for a more significant number of people, and it does not even matter that their effectiveness is marginal. The objective of this work is to provide an analysis of the most recent studies on the treatment of advanced cervical cancer on how the industry conveniently presents the results to give a “magnified” vision of the impact of such results for oncologists and patients as well, specifically about hazard ratio (HR), relative risk (RR) and absolute risk (AR) reductions.
It is increasingly common that the results of randomized cancer clinical trials are presented in hazard risk (HR). The most valuable endpoint in many cancer clinical trials is survival, and the measures derived from Kaplan-Meier survival curves are median survival and 1-, 2-, 3-, 5-year survival estimates. However, most survival differences are assessed after adjustment for covariates, commonly using Cox’s proportional hazards model.10 The HR of the control regimen to the experimental regimen is given by exp[β]. An HR of 0.7 means an HR reduction of 30%. The survival estimates for patients in the experimental arm increase as the HR decreases; however, the magnitude of these survival estimates depends on the survival estimates of the control arm and is not linear.11,12
These are some examples:
OS probabilities with HR 0.8 (HRR 20%). When the 1-y OS probability is 80% in control, the 1-y OS probability in the experimental arm would be 84.5%. When the control arm’s 1-y OS probability is 20%, the corresponding 1-y OS probability in the experimental arm would be 29%.
OS probabilities with HR 0.2 (HRR 80%). When the control arm’s 1-y OS probability is 80%, the 1-y OS probability in the experimental arm is 96%. When the 1-y OS probability is 20% in the control arm, the corresponding 1-y OS probability in the experimental arm would be 72%. A graphical explanation of these concepts are shown in Figure 1.
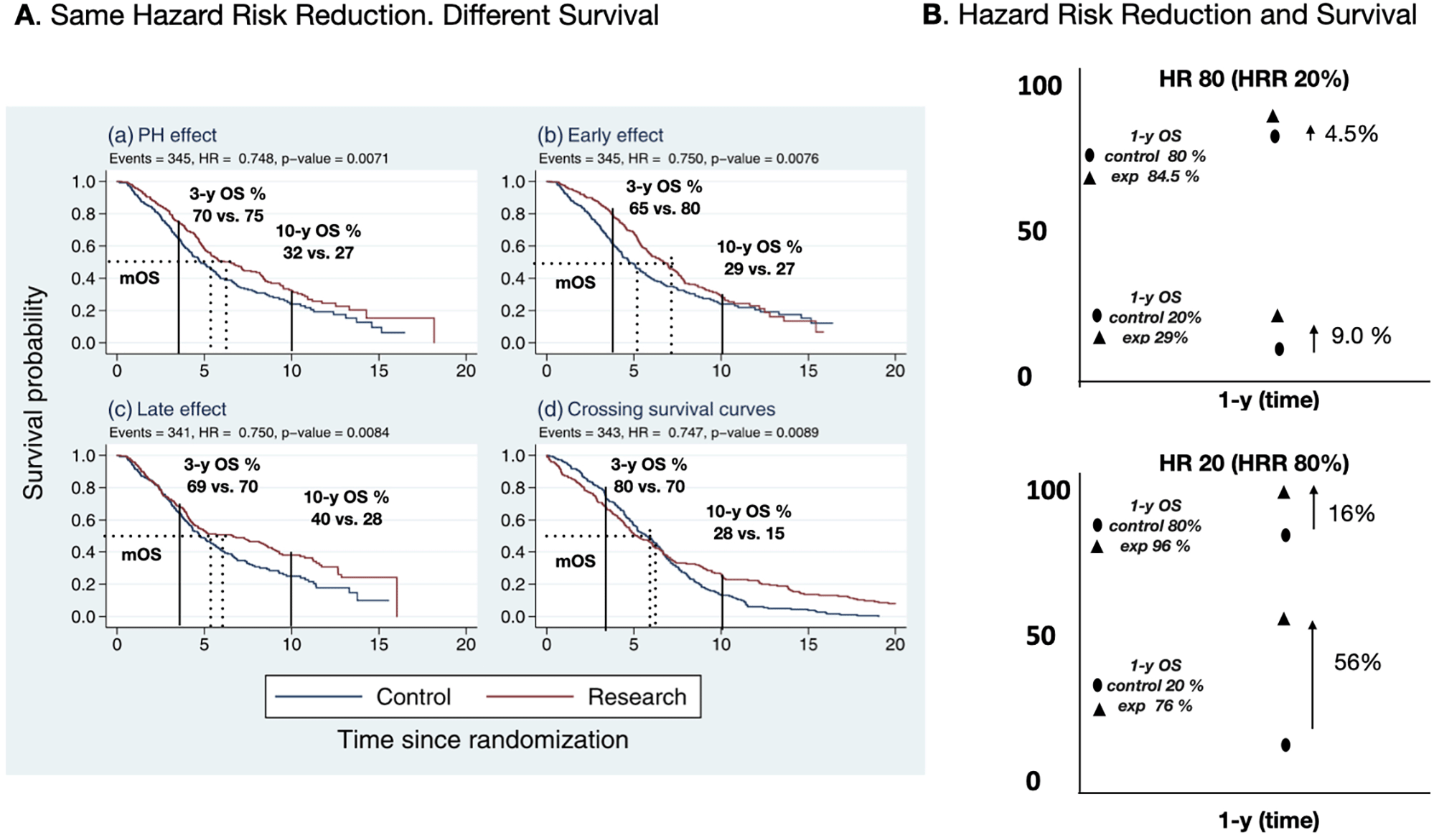
(A) The same HR can have different survival curves which lead to different survival probabilities. The curves can be proportional, can show early effect or late effect and even crossed. (B) Explains how HR reduction results in different survival probabilities depending on the survival probability in the control arms. Panel A is reproduced from Ref 38. Permission was not required.
The HR, however, is a dimensionless value; hence its presentation without the absolute benefit over time provides information of limited value. An HR of 0.5 means that, on average, an individual in the group with the higher HR reaches the endpoint (death in survival analysis) first, or an individual with the lower HR reaches the point last. It should be noted that two survival curves with the same HR may lead to different outcomes. As shown in Figure 1, it is readily appreciated that with similar HR (in the example 0.75), the median OS and 3-y, 5-y, and 10-y OS survival rate estimates are different, illustrating that HR is based on relative rankings and not on actual survival times.11,12
Patients and perhaps some medical oncologists may not have a clear and practical understanding of what relative and absolute risk measures mean. Therefore, the pharma industry takes advantage of this and usually presents the results of cancer clinical trials to obtain a favorable impression from the medical and patient community. Two examples best explaining these concepts are the following:
In a study, 100 cancer patients were treated with a control treatment and 100 with an experimental treatment. two out of 100 patients in the control group died, and in the experimental group, only one died. The RR reduction is 0.50. Two deaths are 100%, and therefore one death is 50%; thus, the RR (RRR) reduction is 50% (0.5).
What is the absolute risk reduction (ARR) from these two theoretical studies? In example 1, the ARR is only 1%. That is: 2% of patients died in the control arm while 1% in the experimental arm (2% minus 1% is 1%). This AR reduction is not statistically significant (95% CI: -0.237-4.37) as the confidence interval crosses zero. In example 2, the ARR is 49% (98% minus 49% = 49%). Here, the 95% CI is 38.82-59.18, which is statistically significant. Based on that example, it is easy to understand that the Relative Risk Reduction (RRR) is how much Risk is reduced in an experimental group compared to the control group, but the RR does not provide any information about the AR of the event occurring. Accordingly, Absolute Risk is one of the most understandable ways of communicating health risks to the general public. RR and AR use binary data (number of deaths in each group) for their calculation. Figure 2 shows these examples.
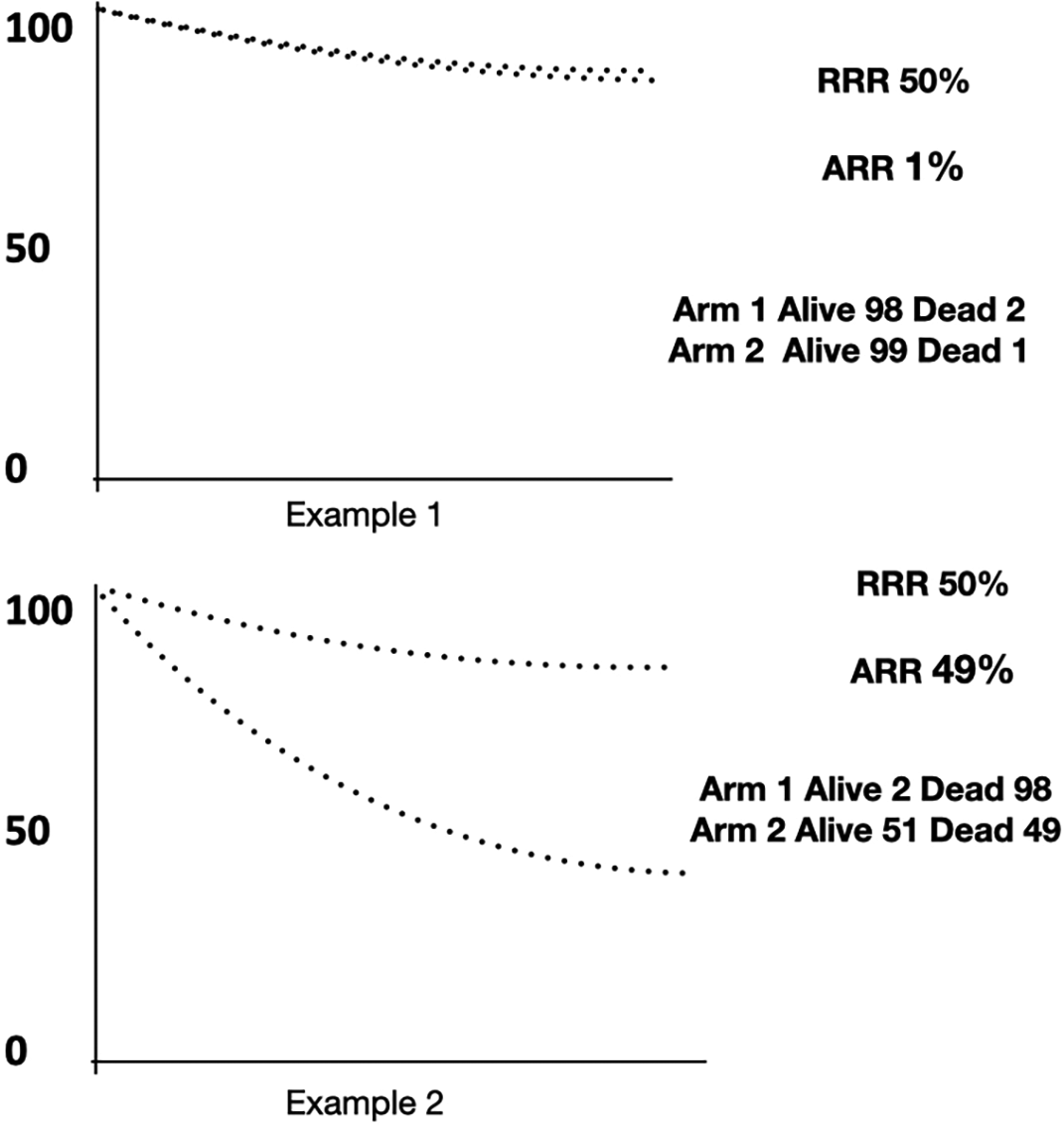
The same relative risk reduction can translate into totally different absolute risk reductions. Curves are imaginary as RR and AR are binary data.
The number-to-treat (NNT) provides an unmistakable idea of a treatment’s impact on the outcome. In the case of an experimental treatment, how many patients need to be treated to prevent an outcome from occurring in an individual (death, for example)? The NNT is simply the reciprocal of the ARR (100/ARR). Consequently, there is an inverse relationship between the effect (ARR) and the NNT. Treatment with an NNT of 10 is superior to an NNT of 50. Returning to the two hypothetical examples, the NNT of the first example is 100 (100/1=100) with a 95% CI: 22.9 to infinity). Because the upper interval goes to infinity, there is no statistical significance. On the contrary, the NNT in example two is 2 (100/49=2.0) (95% CI: 1.7 to 2.6) because the lower interval is above 1 and the upper is not the infinity; the results are statistically significant.13–15 Relative Risk calculations were performed in https://www.medcalc.org/calc/relative_risk.php, and Absolute Risk and NNT in http://araw.mede.uic.edu/cgi-bin/nntcalc.pl?2x2=Compute.
The following case illustrates a real-life example of the enormous discordancy between RR, AR, and NTT. In Israel, 596,618 people were COVID-19-vaccinated, while 596,618 were in the control arm. A total of 32 patients died in the control arm and 9 in the vaccinated arm. The RRR for “Death due to COVID-19” was 71.88%, while the ARR was 0.004%. This translates into an NNT of 25,949 (to vaccinate 25,949 individuals to avoid 1 death). For comparison, vaccination with the Smallpox vaccine in India resulted in the following numbers. The vaccinated and control individuals were 2,377 and 3,147, respectively. Deaths in the vaccinated group were 76, and deaths in the control group were 944. RRR 89.34% and ARR 26.8%. This means that 4 individuals need to get vaccinated to avoid 1 death (NNT 4).16
It is imperative to interpret the Hazard Ratio (HR) meaning correctly. Sasheghy12 stated, “It is a common practice when reporting results of cancer clinical trials to express survival benefit based on the HR from a survival analysis as a “reduction in the risk of death.” Results are commonly presented as follows: drug A reduces the Risk of death by 40% based on an observed survival HR of 0.60. This is a typical but incorrect way of communicating survival benefits. HR should not be interpreted as a RR. If we did not distinguish between HR and RR, the “reduction in risk” (as employed in the publication of clinical trials) implies a durability of the effect in the sense that one is led to believe that for a fraction of the population, the intervention can eliminate the chance of the event to occur. This is not. The “risk reduction” based on HR means a reduction in the speed of the event to happen, not the chances to occur. On the contrary, the “risk reduction” based on RR means that patients have less chance of having the event. Because of that, it is easy to understand why scientific journals and the media highlight the results equating the reduction in HR with the reduction in RR. Moreover, the efficacy expressed in ARR goes unmentioned because AR is always lower than RR. On the contrary, side effects are always presented with absolute numbers or percentages (AR).
The information from early trials (Tighpen 1981, Bonomi, 1985 and Omura 1997),2,3,17 established single agent cisplatin at 50 mg/m2 every 3 weeks as the standard of care. No advantage was observed from adding what is now considered older chemotherapy drugs. There are no significant concerns beyond that studies were not designed or powered for survival parameters but for response rates. Moreover, studies were not analyzed in the intention-to-treat (ITT) principle. Nevertheless, information was considered reasonable to establish single-agent cisplatin at 50 mg/m2 every 3 weeks as the standard of care.
The first indication that a cisplatin-doublet was superior to single-agent cisplatin occurred in 2004 in the GOG169 study. Among 264 eligible patients, 134 received cisplatin, and 130 received cisplatin-paclitaxel. The ORR was higher with the combination (21% vs. 36%). Tough PFS was longer in the combination arm (2.8 vs. 4.8 months, p<0.001), there were no statistically significant differences in median OS (8.8 months vs. 9.7 months). Studies at that time did not report survival with HR. According to the number of deaths reported in each group, The RR was 0.928 (95% CI: 0.845-1.019) for a RR of death of 7.15% and a decrease in ARR of death of 6.45%. Both decreases are not statistically significant. Based on improved responses and PFS, the authors encouraged using this combination for further comparative studies.18
The first randomized trial demonstrating increased OS of a cisplatin doublet against single agent cisplatin was the GOG179 published by Long et al. Originally, this was a 3-arm study, but the MVAC arm was early closed for toxicity. Thus, 294 patients were randomized to cisplatin (145) and cisplatin-topotecan (147). Median PFS was higher in the combination (4.6 and 2.9 months p=0.014), while the mOS were 6.5 vs. 9.4 months (HR 0.76; p=0.017). Hematological toxicity was higher in the combination.4 The RR was 0.908 for a RRR of death of 9% and a decrease in ARR of death of 8.08%. Both decreases are not statistically significant.
The GOG204 study compared the OS among four cisplatin-doublets. Cisplatin-paclitaxel (CP as the reference arm), cisplatin-vinorelbine (CV), cisplatin-gemcitabine (CG), and cisplatin-toptecan (CT). A total of 513 patients were enrolled (118, 117, 119, and 118) in each arm, respectively. The HR for OS survival with the cisplatin-paclitaxel arm as a reference were: 1.15 (95% CI: 0.79 to 1.67) for CV, 1.32 (95% CI: 0.91 to 1.92) for CG; and 1.26 (95% CI: 0.86 to 1.82) for CT. None of these were statistically significantly different. The arms were comparable concerning toxicity except for leucopenia, neutropenia, infection, and alopecia.5 The RR and AR are higher in each experimental arm in all three comparisons but not statistically significant.
This phase III study published in 2014 by Tewari et al.6 randomized 452 patients to chemotherapy with or without bevacizumab at a 15 mg/kg dose. Chemotherapy consisted of six cisplatin-paclitaxel or topotecan-paclitaxel courses every 21 days. The primary endpoint was OS. The results indicated that adding bevacizumab to chemotherapy (either doublet) increased median OS (17.0 months vs. 13.3 months; HR for death, 0.71, p=0.004) in a one-sided test. Higher median PFS and ORR were increased as well in the experimental arm. Patients in the bevacizumab arm had increased incidence of hypertension of grade 2 or higher (25% vs. 2%), thrombo-embolic events of grade 3 or higher (8% vs. 1%), and gastrointestinal fistulas of grade 3 or higher (3% vs. 0%).6 As stated above, RR was 0.935 (95% CI: 0.8043 to 1.0887), p=0.3896, for a RRR of 6.43% (1-0.935)=6.43%. The ARR was 3.96% [-5.05%, 12.98%]. Both RRR and ARR were not statistically significantly different.
This phase III study randomized patients to chemotherapy+bevacizumab plus placebo (309) or pembrolizumab (308 patients). Both schedules are administered for up to 35 cycles. The dual primary endpoints were PFS and OS, each tested sequentially in patients with a PD-L1 >1 and >10. OS was longer in both subgroups >1 and >10, and in the whole population, 24-month estimates of patients alive were 53.0% vs. 41.7%, HR for death 0.64; p<0.001; 54.4% vs. 44.6%; HR, 0.61; p=0.001 and 54.4% vs. 44.6%; HR 0.61; p=0.001 respectively. Median OS in the whole population was higher in the pembrolizumab group, 24.4 vs. 16.3-16.5 months. Grade 3 to 5 adverse events, anemia, and neutropenia were most common in the pembrolizumab group (30.3% vs. 26.9% and 12.4% vs. 9.7%), respectively. Potentially immune-mediated adverse events were 11.4 vs. 2.9%.7
The first randomized phase III trial that demonstrated improved survival in the 2L therapy of advanced cervical cancer therapy was recently published.19 In the trial, patients were assigned to cemiplimab (350 mg every 3 weeks) or the investigator’s choice of single-agent chemotherapy. The primary endpoint was OS. Among 608 patients (304 in each group), the median OS was longer in the cemiplimab group than in the chemotherapy group (12.0 months vs. 8.5 months; HR for death, 0.69; p<0.001). Overall, grade 3 or higher adverse events occurred in 45% of the patients who received cemiplimab and in 53.4% of those who received chemotherapy. Table 1 summarizes these findings.
The treatment of advanced cervical cancer in the 1L has improved, starting with a median of 7.1 months with cisplatin to 24.4 months with the combination of cisplatin-paclitaxel+bevacizumab + pembrolizumab. It must also be noticed that time alone, which may reflect socioeconomic factors, the functioning of health systems, and inclusion criteria in clinical trials, seem to play a role. Though the median OS with cisplatin alone remained unchanged from 1985 to 2004 (7.1-6.5 months), the median OS with cisplatin-doublets (control arms) increased from 9.4 to 13.3 months (3.9 months increase) between 2004 and 2014. The 3.9 months increase observed over these ten years looks the same as the one obtained by adding bevacizumab to the doublet (3.5 months). On the contrary, the addition of pembrolizumab has provided a higher increase (8 months). Regardless of whether someone considers these results excellent, good, average, or bad, the results must be presented in the most objective way possible, avoiding any action to inappropriately influence the medical and patient community so that the treatments are valued in the right dimension of its value.
The GOG43 compared cisplatin doses, and the GOG110 compared cisplatin against cisplatin combined with mitolactol or ifosfamide; they were not designed to test survival but response rate, nor were analyzed in the ITT. The results indicated that there was no advantage of increasing the dose of cisplatin beyond 50 mg/m2, nor that mitolactol or ifosfamide could improve survival over cisplatin. The GOG169 and GOG179 were relatively consistent on the magnitude of survival gains in both PFS and OS of either cisplatin-paclitaxel (GOG169) and cisplatin-topotecan (GOG179), although a statistically significant difference in OS was observed only in the GOG179 trial. Likely, the different proportion of patients with previous chemoradiation or radiation alone could stand for these results. The GOG204 study comparing four-cisplatin doublets with cisplatin-paclitaxel as the reference arm somehow lent support to the benefit of the cisplatin-doublet.5 Interestingly, the GOG179 trial was the first to present survival results in HR; however, the authors did not refer that this combination decreased the Risk of death in the NEJM publication. However, the lay press stated, “there was a 24% reduction in the Risk of dying in patients taking the combination (https://www.cancernetwork.com/view/topotecancisplatin-improves-cervical-cancer-survival).
The GOG240 study evaluating bevacizumab was highly publicized. These are some phrases from the title or the body of the publication taken from the lay press:
“Avastin met its primary endpoint of improving overall survival with a statistically significant 26% reduction in the risk of death for women who received Avastin plus chemotherapy, compared to women who received chemotherapy alone.” https://www.genengnews.com/topics/translational-medicine/avastin-wins-fda-nod-for-advanced-forms-of-cervical-cancer/
“Avastin plus chemotherapy offered a statistically significant 26% reduction in the risk of death” (https://hospitalpharmacyeurope.com/news/editors-pick/first-new-treatment-authorised-in-a-decade-for-advanced-cervical-cancer-patients-in-the-uk/)
“Avastin: first molecule in nearly a decade for metastatic cervical cancer. There was a 26% reduction in the risk of death when bevacizumab was combined with chemotherapy.” (https://www.ajmc.com/view/avastin-first-molecule-in-nearly-a-decade-for-metastatic-cervical-cancer)
The NEJM publication6 that served for the above (paragraphs taken from the abstract) states the following:
The addition of bevacizumab to chemotherapy was associated with increased overall survival (17.0 months vs. 13.3 months; HR for death, 0.71; 98% CI: 0.54 to 0.95; p=0.004). Bevacizumab, as compared with chemotherapy alone, was associated with an increased incidence of hypertension of grade 2 or higher (25% vs. 2%), thrombo-embolic events of grade 3 or higher (8% vs. 1%), and gastrointestinal fistulas of grade 3 or higher (3% vs. 0%).
What would the above look like if HR is not equated to RR and if AR for efficacy and RR for toxicity were mentioned?
Efficacy, as stated:
Median overall survival: 17.0 months vs. 13.3 months. HR for death, 0.71; p=0.004.
Efficacy, unmentioned:
Relative Risk: 0.935 (95% CI: 0.8043 to 1.0887), p=0.3896. RRR (1-0.935)=6.43%. This reduction is not statistically significant.
Absolut Risk Reduction 3.96% [-5.05%, 12.98%]. The 95% CI crosses zero, implying that the ARR is insignificant. The new therapy may increase Risk.
Hypertension >2 (25% vs. 2%). Absolute Risk increased by 23%. Relative Risk Increase 1,493%.
Thromboembolic events >3 (8% vs. 1%). Absolute Risk increased by 7%. Relative Risk Increase 593%.
Gastrointestinal fistulas >3 (3 vs. 0%) Absolute Risk increase of 3%, Relative Risk Increase 1,343%.
It is readily appreciated that data presentations are aimed to positively impact the audience, establishing a 29% lower risk of dying, assuming that the HR is the same as the RR, but it is not. The data indicate that at a median follow-up of 20.8 months, 140 deaths occurred in the chemotherapy control arm (219 patients) and 131 in the chemotherapy plus bevacizumab arm (220 patients). It means that patients who receive the experimental treatment have a 29% less probability on average of reaching death; that is, the speed for the outcome is reduced and therefore results in a statistically significantly higher median survival (17 vs. 13.3 months). However, the RRR of dying is only 6.43%, and the ARR of dying is 3.96%, both cases without statistical significance. The minor ARR translates into an NNT of 22. The survival curves overlap at 30 months of follow-up. On the contrary, if toxicity were expressed in terms of RR and not AR, any patient would hardly accept risks higher than 1000% of suffering serious adverse events if the chances of being alive at 20.8 months are only 3.96% less.
The KEYNOTE-8267 study evaluating pembrolizumab also was highly publicized. These are some phrases taken from the media.
“Merck’s KEYTRUDA® (pembrolizumab) plus chemotherapy with or without bevacizumab reduced Risk of death by one-third versus chemotherapy with or without bevacizumab as first-line treatment for persistent, recurrent or metastatic cervical cancer (https://www.merck.com/news/mercks-keytruda-pembrolizumab-plus-chemotherapy-with-or-without-bevacizumab-reduced-risk-of-death-by-one-third-versus-chemotherapy-with-or-without-bevacizumab-as-first-line-treatment/)
“The data showing a 36% reduction in the risk of death are compelling” (https://www.pharmatimes.com/news/fda_clears_keytruda_for_first-line_cervical_cancer_1380837)
“Adding the checkpoint inhibitor pembrolizumab (Keytruda) to standard chemotherapy - with or without bevacizumab - resulted in about a one-third reduction in the risk of death compared with chemotherapy alone.” (https://cancerletter.com/clinical-roundup/20210924_8c/#:~:text=Keytruda%20%2B%20chemo%20reduces%20risk%20of%20death%20by,or%20metastatic%20cervical%20cancer%20-%20The%20Cancer%20Letter).
The NEJM publication7 that served for the above (paragraphs taken from the abstract) states the effect of pembrolizumab.
Overall survival at 24 months was 50.4% and 40.4% (HR, 0.67; 95% CI: 0.54 to 0.84; p<0.001). Though unmentioned in the abstract, the body of the manuscript and figures appear as “The hazard ratio for death.” The most common grade 3 to 5 adverse events were anemia (30.3% in the pembrolizumab group and 26.9% in the placebo group) and neutropenia (12.4% and 9), respectively.
What would the above look like if HR is not equated to RR, and if absolute risks for efficacy and relative risks for toxicity were mentioned?
Efficacy, as stated:
Overall survival at 24 months was 50.4% and 40.4% (HR for death, 0.67; p<0.001. Median OS was 24.5 months for pembrolizumab, ranging from 16.3 to 16.5 months for placebo.
Efficacy, unmentioned:
The number of patients that died in each arm unexpectedly was not disclosed, only the total number of deaths which was 312. This number is shown in Figure 3D and Table S1 of the original publication. Because of that, the RR and AR cannot be calculated; however, estimating the number of deaths in both groups from the 50.4% and 40.4% alive in each arm as the authors refer, the number of dead patients would be 153 and 185 respectively for a total of 338. With these death numbers, the following is calculated (it should be noticed that 338 does not correspond to 312).
Relative Risk: 0.829 (95% CI: 0.717 to 0.959), p=0.0115. Relative Risk Reduction (1-0.829)=17%. This reduction is statistically significant.
Absolut Risk Reduction 10.20% (95% CI: 2.38% 18.0%). This reduction is statistically significant as well.
Toxicity: Expressed as Absolute Risk and Relative Risk.
Anemia grade 3-5 (30.3% vs. 26.9%). Absolute Risk increased by 3.4% (NS). Relative Risk increase of 112% (NS).
Neutropenia grade 3-5 (12.4) vs. 9%). Absolute Risk increased by 3.4% (NS). Relative Risk increase of 127% (NS).
Potentially Immune-Mediated Adverse Events (suppl data). Any event grade 3-5 (11.4% - 2.9%).
Absolute Risk increased by 8.5%, p=0.0001 (significance not stated in the publication). Relative Risk increase of 395%, p=0.0002.
Among all the randomized trials in advanced cervical cancer in the 1L setting, pembrolizumab has shown the highest increase in the median OS, which is 8 months. Nevertheless, the data presented in scientific journals and press do misinterpret the meaning of HR and equate it with decreased Risk of death when in fact, this indicates that patients who receive the pembrolizumab have a 33% less probability on average of reaching death, that is, the speed for the outcome is reduced and therefore results in a higher median survival (24 vs. 16.4% months). While the exact RR cannot be determined because the number of dead patients in each arm was not provided, the RR calculated from the survival curve percentages indicate the patients receiving the drug have 17% less Risk of dying at the follow-up reported. In terms of absolute risk reduction, the number is 10.2%, which indicates that only 10 patients are NNT to see a reduction of an event. On the contrary, while it is not incorrect to present toxicity results in absolute percentages, it must be considered that oncologists and patients need to have well-balanced information concerning the benefits and risks of any therapy. Figure 3 provides an overview of these 3 trials using relative, absolute and NNT in addition to month increases in median OS.
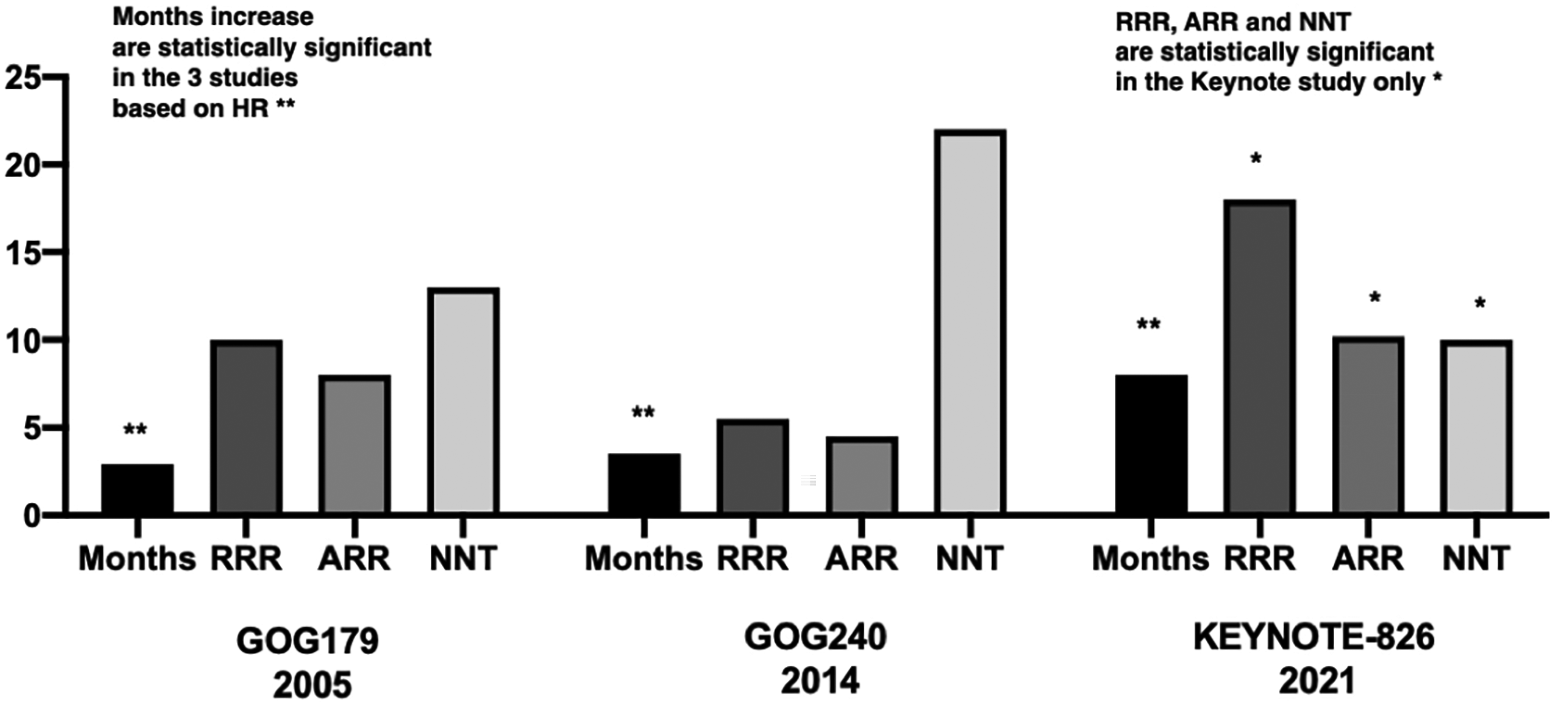
At left are the bars for months increases which all were statistically significant, expressed as HR. It must be noted that calculated RR, AR and NTT are only significant in the Keynote trial.
The EMPOWER- Cervical 1/GOG-3016/ENGOT-cx9 was recently published.25 The study results were also highly publicized in the lay press. These are some phrases taken from the media:
“Cemiplimab reduces the risk of death by 31% vs. chemotherapy in advanced cervical cancer.” (https://www.onclive.com/view/cemiplimab-reduces-risk-of-death-by-31-vs-chemotherapy-in-advanced-cervical-cancer).
“Treating recurrent cervical cancer with cemiplimab reduces the risk of death by 31% compared with single-agent chemotherapy”(https://conferences.medicom-publishers.com/specialisation/oncology/cemiplimab-boosts-survival-in-recurrent-cervical-cancer/).
“Cemiplimab Impresses in a Phase 3 Trial of Cervical Cancer, Regulatory Submission Expected: Treatment with cemiplimab (n=304) reduce the Risk of death by 31% compared with chemotherapy” (https://www.globenewswire.com/news-release/2021/03/15/2192446/0/en/Phase-3-trial-of-Libtayo-cemiplimab-monotherapy-in-advanced-cervical-cancer-stopped-early-for-positive-result-on-overall-survival.html).
The NEJM publication19 states the following:
A total of 608 women were enrolled (304 in each group). In the overall trial population, median overall survival was longer in the cemiplimab group than in the chemotherapy group (12.0 months vs. 8.5 months; hazard ratio for death, 0.69; p<0.001). Overall, grade 3 or higher adverse events occurred in 45 % of the patients who received cemiplimab and in 53.4% of those who received chemotherapy.
What would the above look like if HR is not equated to RR, and if absolute risks for efficacy and relative risks for toxicity were mentioned?
Efficacy, as stated:
Median overall survival at 12 vs. 8.5 months (HR for death, 0.69; p<0.001).
Efficacy, unmentioned:
Relative Risk: 0.872 (95% CI: 0.775-0.980), p=0.0224. Relative Risk Reduction (1-0.829)=12.8%. This reduction is statistically significant.
Absolut Risk Reduction 8.88% 95% CI: 1.33% - 16.43%]. This reduction is statistically significant as well.
Toxicity: Expressed as Absolute Risk and Relative Risk.
Overall grade 3 or higher (45% vs. 53.5%). Absolute Risk increased by 8.4% (NS) (higher in the control group). Relative risk increase of 15% (NS) (higher in the control group)
Sponsor-identified immune-related Adverse Events in >1% (suppl. data)
Grade 3 or higher (5.3% vs. 0.7%). Absolute Risk increased by 4.6%. Relative Risk increase of 773%, p=0.006
After many years of studying 2L therapy for advanced cervical cancer, cemiplimab has been shown to increase the median survival time from 8 to 12.5 months which is a step ahead. The presentation of the results, however, suffers from the same problem that the HR decrease is misinterpreted as reducing death risk. The results indicate that cemiplimab decreases by 31% the Risk of reaching death. Because of that, the median survival time increases by 4.5 months. What is not said is that, indeed, cemiplimab reduces the relative Risk of death by 12.8% and by 8.8%, both of which are statistically significant. The presentation of toxicity, while adequately presented in the body of the manuscript regarding overall grade 3 and higher, the higher immune-related toxicity is presented in supplementary information only.
Beyond data on how the results of cancer clinical trials are presented, the design of these must be rigorous and well-conducted. There must be a careful balance between the best design to demonstrate the superiority of an experimental treatment as cleanly and clearly as possible and the best treatment for a patient. In no case any trial maneuver must be above the patient’s interest and allow a potentially inferior control arm.20
There are no significant concerns regarding the first randomized trials until the GOG204 study. This is not so for the GOG240 testing bevacizumab. The chemotherapy control arm with either doublet is adequate. It is remarkable, however, that in the protocol design, no therapeutical action was contemplated beyond study termination (progression, toxicity, or patient consent withdrawal). Current and past (when the study was conceived) NCCN guidelines contemplate using second-line therapy as standard. Among therapeutical options are single-agent therapy (albumin-bound paclitaxel, docetaxel, fluorouracil, capecitabine, gemcitabine, ifosfamide, irinotecan, mitomycin, and vinorelbine).
A recent study characterized the patient experience, treatment patterns, and clinical outcomes of patients who initiated 2L therapy for advanced cervical cancer in a US community oncology setting. Among 130 patients identified (more than 60% had ECOG score of 0-1), 46% received single-agent chemotherapy, 15% bevacizumab, 19% pembrolizumab (this agent was not approved by the time the GOG240 study was done), and 34.6% combination chemotherapy. Moreover, 58 of these 130 patients (44.6%) received third-line therapy.21 Another study from the UK reported that among 75 patients, 53 (70.7%) received 2L therapy. The most common second-line therapy was weekly paclitaxel (28.3%), carboplatin-based chemotherapy (24.5%), targeted agent monotherapy within clinical trials (22.6%), docetaxel-based chemotherapy (13.2%), topotecan (9.4%), and gemcitabine (1.9%).22 It is easy to understand that while the PFS endpoint could not be affected by adding 2L therapy, the OS endpoint would potentially do. This is a clear example of how the sponsor’s interest was above the patient’s. One wonders if patients were informed of this when they entered the study. Accordingly, Figure 1 (of the original publication) of the flow chart of the study reveals that only 51 (22.6%) and 33 (24.5%) of chemotherapy and chemotherapy-bevacizumab crossover receive salvage therapy. None of the “approved” 2L therapies were employed beyond the study drugs. It is logical to question should patients in both arms have received approved 2L therapies; the OS could have changed. On the other hand, it seems that currently, it is forgotten that combination versus sequential chemotherapy results in similar survival times in breast, lung, and ovarian cancer23–27 and even single-agent versus combined targeted drugs in renal cancer.28 Bevacizumab is an effective drug, but it is expensive and not devoided of toxicity. Should the trial have an additional arm administering chemotherapy following bevacizumab at progression had similar survival than the concurrent combination? Patients and health systems could benefit if this were the case by reducing the amount of bevacizumab used without jeopardizing survival.
The KEYNOTE-826 has similar observations regarding the use of 2L. The use of new antineoplastic therapy was explicitly prohibited. Even more, as referred above, when the protocol began, single-agent pembrolizumab was already approved for its use as one of the standards of 2L therapy. The scenario is similar to that of bevacizumab. Had the use of 2L therapy could have changed to study outcome regarding OS? Ideally, a third arm using concurrent chemotherapy and pembrolizumab at progression would have resulted in similar survival to the concurrent treatment. While a third arm, if proven equally effective, would benefit patients and health systems, it would reduce the product’s profitability.
In the cemiplimab (EMPOWER- Cervical 1/GOG-3016/ENGOT-cx9) study, salvage therapy in the control arm could be even more debatable. It has been described the multiple single-agent options for the 2L therapy and that a substantial proportion of patients in real-world practice receive 3L therapy.22 The protocol specified that the investigator could use options “reflecting the availability of drugs in different regions of the world,” these were pemetrexed, topotecan, irinotecan, gemcitabine, and vinorelbine (all other drugs excluded). Of note, a patient initially randomized to chemotherapy may be considered for resumption of the same chemotherapy they received in the treatment period. This is particularly questionable as every oncologist would hardly reuse the same drug to which the patient progressed.
Moreover, single-agent paclitaxel, bevacizumab, and pembrolizumab (options marked for 2L therapy by the NCCN, were excluded. Beyond any theoretical support for not allowing the use of salvage chemotherapy in both arms and limiting treatment options in the control arm, it is clear that the interest in having a “clean and solid” statistical result was superior to the patient’s interest. Accordingly, the same question arises, should the study have allowed a “truly-investigator choice” of therapy (including any other single agent, chemotherapy agent, bevacizumab, or pembrolizumab), could the results be different? Should a third arm using truly investigator-choice chemotherapy followed by cemiplimab at progression could change study results in favor of patients and society by reducing the amount of cemiplimab use without compromising survival?
There have been advances in the treatment of advanced cervical cancer. Here we critically analyzed how the results are communicated to the medical oncology community, patients, and society, particularly for the two previous randomized studies in 1L. In the author’s opinion, the communication of these clinical trial results is somehow unbalanced. Medical journals and lay press tend to overestimate the benefits and underestimate novel therapies’ risks.
The main issue has to do with how survival results are communicated. As stated above, modern oncological trials use the HR as a measure to prove differences in survival statistically, but the HR should not be interpreted as a RR. The main difference between both is that RR is the Risk of dying without considering the time factor, while HR is the Risk of dying considering the time factor. Because of that, HR is reported most commonly in time-to-event analysis or survival analysis. If we did not distinguish between HR and RR, the “reduction in risk” (as employed in the publication of clinical trials) implies a durability of the effect in the sense that one is led to believe that for a fraction of the population, the intervention can eliminate the chance of the event to occur. This it is not. The “risk reduction” based on HR means a reduction in the speed of the event to happen, not the chances to occur. On the contrary, the “risk reduction” based on RR means that patients had less chance of having the event.
The topic of patient expectations regarding treatment results, meaning gaining extra months of life, “delaying the time of death,” or having chances to avoid the event (death), is really complex and not further discussed here.29–32 However, at least one study states that patients maintain hope when they receive truthful prognostic and treatment information, even when the news is bad.33 Patients need to be communicated most straightforwardly and accurately as possible about the benefits of cancer treatments. The study’s results must indicate how the study drug can decrease the Risk over time (median survival time) (HR), to what extent the Risk of death is decreased, and, perhaps, the most important and often neglected is communicating absolute Risk. It means the absolute reduction in Risk for the patient to die. The authors are far to be experienced statisticians, but the pertinence of using or not using the measures of RR and AR at least merits debate. These differences in the meaning and interpretation of clinical results underlie why efficacy is always presented in relative Risk but toxicity in absolute Risks.
Figure 3 shows the results of the three main randomized trials in the 1L management of advanced cervical cancer. The first bar represents the months gained in each study which are 2.9 (GOG179), 3.5 (GOG240), and 8 (KEYNOTE-826) months, while the corresponding HR reductions are 24% (0.76), 23% (0.77), and 35% (0.65). In the three cases, the HR reductions are statistically significant. The months gained correspond with the magnitude of risk reduction in terms of HR. Consequently, it is easier to understand that HR reductions are translated to months gained. This is routinely presented, except that HR reductions are treated as RR.
On the other hand, if RR were informed, one could observe that RRR is lower in the GOG179 (10%) and GOG240 (5.5%) as compared to the KEYNOTE-826 (18%). The statistical significance was only reached in the KEYNOTE-826 trial. This means that patients treated in the KEYNOTE-826 in the pembrolizumab arm have 18% less relative Risk of dying than patients not receiving the study drug. This picture behaves the same concerning the AR. Patients receiving pembrolizumab have a 10.2% less absolute risk for death which is statistically significant. Accordingly, one needs to treat 13 patients with cisplatin-topotecan to avoid one event (death), 22 patients with chemotherapy-bevacizumab to avoid 1 death, and finally, only 10 patients with chemotherapy-bevacizumab-pembrolizumab in order to prevent death (in this case, the 95% CI: is 5.6-42), therefore is considered statistically significant. The presentation of the results using these measures provides a more informative picture demonstrating that the GOG240 provides less RR and AR reductions and, therefore, the need to treat more patients to observe the effect. On the other hand, the study providing the higher benefit in these four parameters is the KEYNOTE-826 study.
There is a growing interest in discussing how the industry influences the design, analysis writing, and publication of clinical cancer trials. It seems that it has much more weight in designing studies to demonstrate spotlessly and soundly that the drug of study is superior, no matter if patients are not allowed to receive the full options of “standard” therapies or be allowed to crossover or not at progression. If a drug is already approved for the 2L, there is no reason (other than the industry interest) not to offer it to patients progressing to the first line. It is argued that if protocols allow for crossover, the study outcome can be negative because of this “contamination.” Do these maneuvers of the sponsor have anything to do with doing the best for patients?
In the recent past, it was common to perform studies to optimize treatment for a patient. Specifically, combination versus sequential therapies. It has been observed that most of the time, sequential therapies may result in equal survival. The need to add additional arms to prove that the sequential use of novel therapies has similar efficacy in terms of survival must not be underestimated. It is understandable, however, that most, if not all, of recent standard-changing trials are sponsored and designed by the pharmaceutical industry, and its interest is to profit as much as possible. Why should sponsors be interested in demonstrating that sequential is the same in terms of survival that combination if sales would decrease with the sequential approach?
At the individual level, the high prices of novel cancer drugs result in poor patient affordability. Even those who can afford these treatments can suffer from financial toxicity, increasing the Risk of death.8 At the societal level, the unsustainability of the current drug development and research model is widely discussed, and potential solutions have been proposed.34–37 To overview the magnitude of the problem, let us provide an example of what could represent treating advanced cervical cancer patients with the “standard” platinum-paclitaxel-bevacizumab-pembrolizumab in a middle-high income country. In Mexico, the Federal Budget for Health in 2022 was 9,697 billion USD (at an exchange rate of 1 USD/20 Mexican Pesos (https://www.pef.hacienda.gob.mx/work/models/aVbnZty0/PEF2022/kgp8l9cM/docs/12/r12_ppcer.pdfhttps://www.pef.hacienda.gob.mx/work/models/aVbnZty0/PEF2022/kgp8l9cM/docs/12/r12_ppcer.pdf). In 2019 it was reported that in Mexico, 4,800 women died of cervical cancer (cdn.who.int/media/docs/default-source/country-profiles/cervical-cancer/cervical-cancer-mex-2021-country-profile-es.pdf?sfvrsn=8a0b4124_38&download=true). If we round up this number to 5,000 and assume that all these women are treated, the cost per patient (retail price) for bevacizumab and pembrolizumab would be 883 billion USD, representing around 9.1% of the total health budget. For any health system, this is nonsense.
The communication of the results of cancer clinical trials by medical journals and lay press needs to be balanced. In particular, the correct interpretation and communication of Hazard Risk Reduction meaning are fundamental. It should not be equated with Relative Risk Reductions because it can be misleading to patients and clinicians. Prolonging survival does not mean averting the Risk of death. HR means a reduction in the event’s speed, not the chances of occurring.
Ideally, ARR and NNT should always be used because it is the simplest form to communicate the treatment effects straightforwardly. After all, patients have the right to know the most accurately possible what to expect from any treatment. One can argue against the use of these statistical parameters for their simplicity. However, we must not forget that medicines, if they are marvelous or miraculous, may not require statistical analysis. The need to use more complex and sophisticated statistics parallels their limited effectiveness. It seems all is about demonstrating that the slightest benefit on survival parameters reaches statistical significance to register and commercialize novel drugs. Figure 4 shows data needed to have a more informative on the value of any therapy tested.
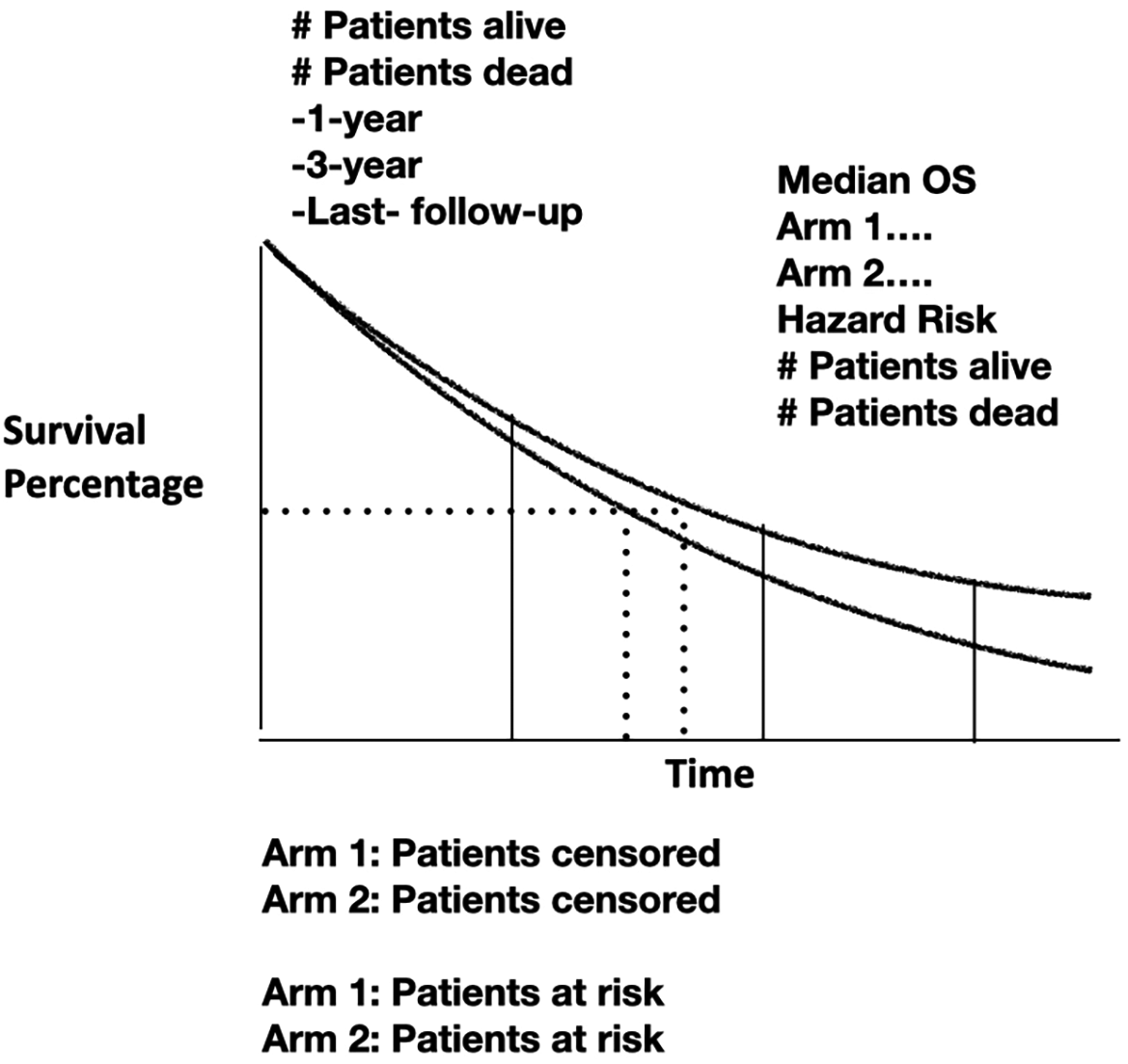
Hazard Risk must be accompanied by binary outcomes, alive or death at different times to calculate risks. Censored patients must be included but also the detailed censoring plan in methods section.
On the other hand, the interest of the patients must always prevail over the industry’s interest in having a “solid and clean” result in clinical trials. Patients must be offered all available treatment interventions that have proven efficacious and to allow crossover when the experimental agent being evaluated is already approved as a salvage therapy. These two actions must be done even if they could confound the clinical trial results. Every effort should be made to embark on clinical trials prioritizing the optimization (in terms of efficacy, toxicity, and cost) of novel drugs, even if this affects the industry’s profitability. Sadly, the price of novel cancer treatments is such that no one health system can sustain the current clinical cancer research and commercialization model that uses the “willing-to-pay” approach to establish drug prices.
We all want to achieve real progress in cancer control, but we must be honest about how well we are doing and avoid using statistics conveniently to present the facts more attractively and using superlative adjectives. Exaggerating progress could be offensive to anyone who has lost someone to cancer or is currently fighting cancer. It can be argued that the expectation of cancer treatment advances has shifted from a definitive cure to disease control. Under this statement, relative, absolute, and NNT are undoubtedly useless. However, by accepting this, we impose ourselves limits for achieving significant strikes on cancer.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (1)