Keywords
Anopheles squamosus, understudied malaria vector, Africa, Zambia
This article is included in the Genomics and Genetics gateway.
Anopheles squamosus, understudied malaria vector, Africa, Zambia
Anopheles squamosus (Theobald, 1901; Figure 1) can be found across Africa1 and is of particular relevance to public health due to its implication in the spread of residual malaria cases. Anopheles squamosus is one of the most abundantly caught anopheline species in malaria vector surveillance studies in southern Zambia. However, it is understudied species because of its exophilic and zoophilic behaviours.2,3 Though they are predominantly associated as a zoophilic species, they have been discovered to have high anthropophily in southern Zambia.4 Additionally, there has been the detection of Plasmodium falciparum sporozoite and DNA, a causal agent of human malaria, in An. squamosus.2,5

A and B have been reproduced with permission from Dr. Rebekah Kading (Colorado State University).21 C has been reproduced with permission from Walter Reed Biosystematics Unit (WRBU).22
Unfortunately, there are two key barriers to pursuing the rigorous investigation of the epidemiologically important traits of this vector, such as host choice, biting behaviours, and dispersal capacity. First, An. squamosus is morphologically indistinguishable from An. cydippis at the adult stage. Although they are morphologically distinct as larvae, larvae are often difficult to locate in abundance. There are numerous examples of sympatric Anopheles sibling species expressing drastic differences in insecticide resistance6 or host choice.7,8 These differences make species confirmation critical to assessing and mitigating malaria transmission risk. Second, there is limited genetic information (173 sequences total in GenBank as of August 2022) for An. squamosus, most (N=166; 96%), are partial sequences of the mitochondrial cytochrome c oxidase subunit I (COI) gene. ITS2 sequences are better at differentiating species within a complex than COI sequences9 but existing ITS2 primers do not typically work on An. squamosus and the absence of sequence data for this region prohibit the design of functional diagnostic PCR primers.
To overcome these two barriers and advance investigative efforts aimed at this widespread, yet neglected malaria vector, we carried out the first Illumina high-throughput sequencing of this species.
The An. squamosus sample used for the genome sequencing was collected in Chidakwa near Macha, Zambia (utm-x: 0478202, utm-y: 8184394) using a CDC light trap placed outdoors near a goat pen. Samples were frozen after collection at –20°C until DNA extraction. DNA was extracted using a magnetic bead-based protocol optimized for mosquito DNA for Next-generation sequencing.10 The head and thorax were dissected from the sample and hydrated in nuclease-free water for 1 hour at 4°C. Tissues were then removed from the water and homogenised in a mixture of 2 μL Proteinase K (100 mg/mL) and 98 μL PK Buffer in a 1.5 mL Eppendorf microcentrifuge tube (add company name), followed by incubation at 56°C for 2 hours. The lysate was transferred to a new 1.5 mL microcentrifuge tube and mixed with a MagAttract Mix consisting of 100 μL isopropanol, 100 μL Buffer AL, and 15 μL MagAttract Suspension G (Qiagen, Hilden, Germany). The mixture was incubated at room temperature for 10 minutes and occasionally vortexed to ensure that the magnetic beads were evenly dispersed. The microcentrifuge tube containing the lysate was then moved to a magnetic bead separator until the liquid appeared clear. After a series of ethanol washes of magnetic beads, DNA was eluted from the beads with 100 μL AE Buffer and stored at −20°C until library preparation. The library preparation was completed using the QIAseq FX UDI kit (Qiagen, Hilden, Germany) using 20 ng genomic DNA as input for the protocol as previously described.11
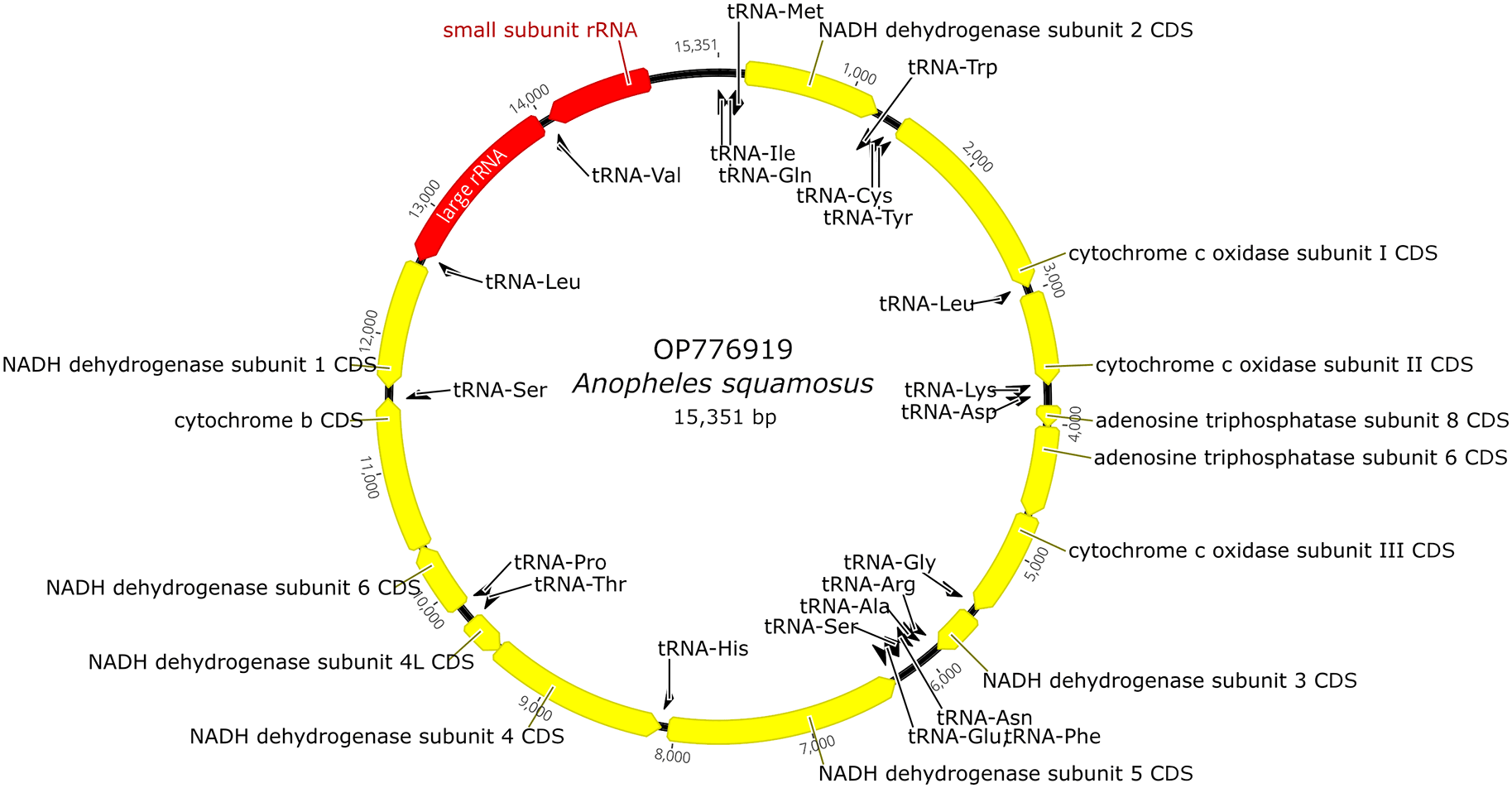
Raw sequencing reads were trimmed using fastp (RRID:SCR_016962) version 0.20.1.12 Mitogenome (Mt) contig was assembled using NOVOPlasty (RRID:SCR_017335) version 4.3.1.13 Automatic annotation of mitogenome was conducted with the MITOS website14 using the invertebrate genetic code for mitochondria under default settings. Some automatic annotations were not consistent with typical Anopheles mitochondrial gene start and/or end positions. Manual adjustments were made to inconsistent automatic annotations by shifting the start and end positions to match existing Anopheles mitochondrial gene annotations found in GenBank. Annotation information was also deposited to the GenBank with the genome sequence. The full genomic map is provided in Figure 2.
Phylogenetic analysis was conducted using the mitogenome sequences of seven Anopheles species and one Aedes species as an outgroup in. The Jukes-Cantor model was used to calculate the pairwise genetic distances and the neighbour-joining method was used to build the phylogenetic tree in Geneious Prime (RRID:SCR_010519) 2022.02 (Biomatters, Auckland, New Zealand)15 (free alternative, AliView).
Draft genome assembly was conducted using MaSuRCA (RRID:SCR_010691) version 4.0.916 in order to find a contig containing Internal transcribed spacer 2 (ITS2) sequence. Basic local alignment search tool (BLAST) (RRID:SCR_004870) was used for the resulting contigs to locate contigs with highest similarity with only An. squamosus ITS2 sequence available on GenBank (accession number MK592071).
We yielded 105 million reads from sequencing a single An. squamosus sample. Of these, 238,740 reads were used to assemble mitochondrial genome. Draft genome assembly using MaSuRCA produced 58,252 scaffolds with the total size of scaffolds of 350Mbp. N50 scaffold length was 21,439bp. Among these contigs, we identified one contig containing ITS2 region (GenBank accession number OQ241725), which was 1,223 bp long.
The length of the An. squamosus Mt (GenBank accession number OP77691923) was 15,351 bp and the percentage A+T was 77.9% (Figure 2). The average A+T percentage of eight other anopheline species was 77.7% (±0.61 SD). The length of this mitochondrial genome was a similar length to other anopheline species that have been deposited in GenBank, with the average of the eight species compared in this analysis being 15,363 bp. The content for this mitochondrial genome includes two ribosomal RNAs, 22 transfer RNAs, and 35 protein-coding genes. The cytochrome c oxidase I (COI) fragment spanning 1,462-2,132 bp of An. squamosus sequence had 97.7% (±4.27 SD, N=9) similarity to the COI sequence of An. squamosus deposited in GenBank.
In the phylogenetic analysis (Figure 3), the closest match to the whole mitogenome sequence of An. squamosus was the African malaria mosquito An. gambiae (GenBank accession number L20934), with 91.5% sequence similarity. This comparable sample was identified as An. gambiae and published in 1993 before An. gambiae were separated into two species: An. gambiae and An. coluzzii.17 Nevertheless, previous studies suggest that mitogenome sequence alone is not sufficient to distinguish An. gambiae s.s. from An. coluzzii.18,19
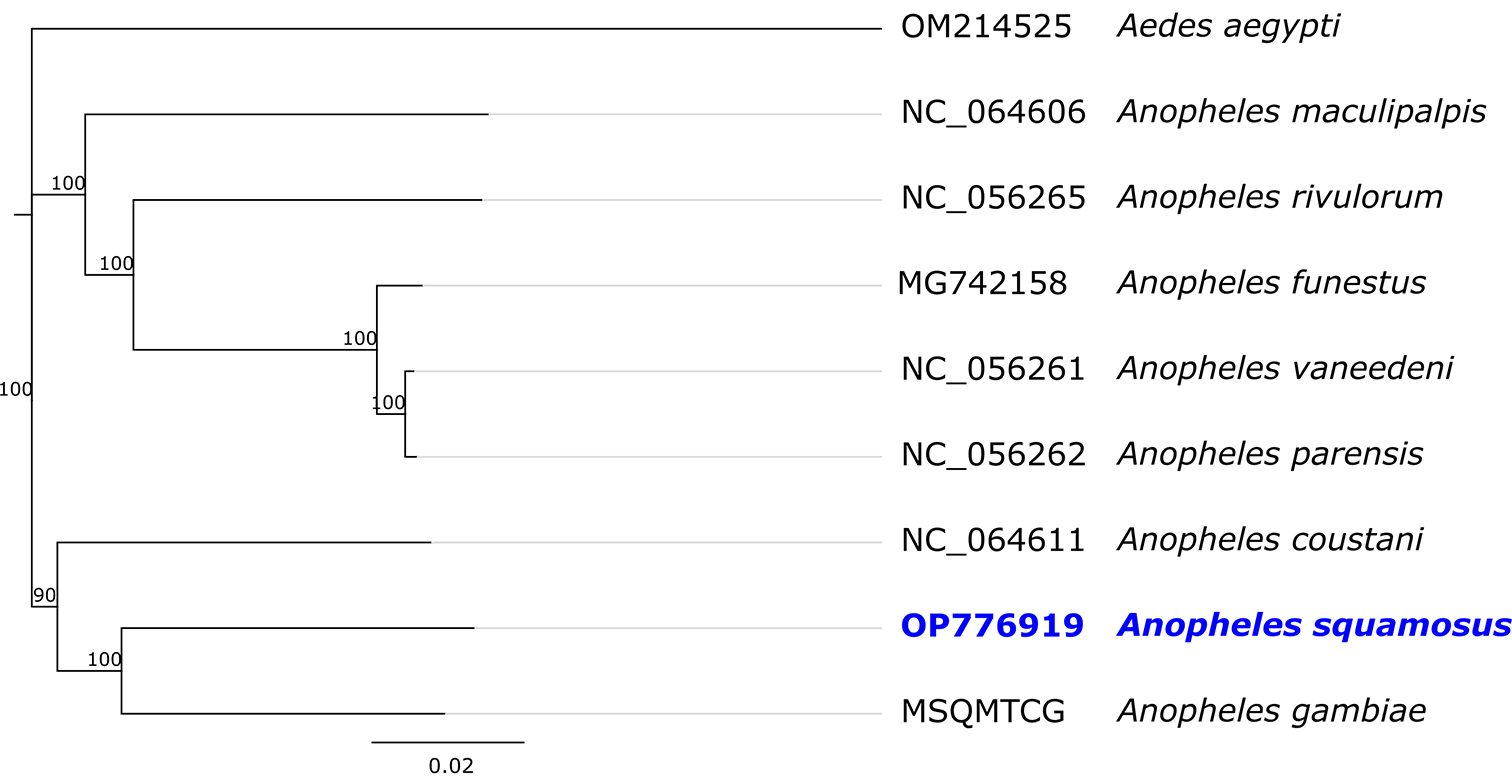
Species names are provided next to the GenBank accession numbers. Numbers at nodes indicate bootstrap values out of 100 replicates. Aedes aegypti was considered as an outgroup. The scale bar indicates relative nucleotide difference (0.02=2% nucleotide difference).
This study provides a critical genomic resource for research of this understudied malaria vector. Our short reads sequencing data was not sufficient to assemble high-quality reference genome and revealed the need for alternative long-read sequencing technology for a high-quality genome assembly. However, we provided a key ITS2 region data that researchers can develop a low-cost molecular diagnostic assay to identify species. Currently available ITS2 primers for anopheline species identification typically does not produce a PCR amplicon, which is one of the major roadblocks in carrying out surveillance and research of this species. We identified the ITS2-containing contig (GenBank Accession number OQ241725) that could be used for new primer design that would amply the ITS2 fragments more reliably for An. squamosus. Our genome sequence data could be used for further variant identification once high-quality reference genome become available for An. squamosus. The mitogenome sequence could also be used to identify phylogenetic relationship within and between related species and infer gene flow/dispersal.9,20
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)