Keywords
Mizoribine, Lupus Nephritis, randomized controlled trial, Cyclophosphamide
This article is included in the All trials matter collection.
Mizoribine, Lupus Nephritis, randomized controlled trial, Cyclophosphamide
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by the production of auto-antibodies and symptoms that result from the damage of multiple organs.1 Among the many organ complications, SLE with kidney involvement, known as lupus nephritis (LN), accounts for most of the morbidity and mortality in SLE patients.2–5 In particular, 10 to 30% of patients with LN develop end-stage renal disease (ESRD) within 15 years after diagnosis,6–10 and ESRD is a major cause of death in SLE patients.11,12
Intravenous cyclophosphamide (IVCY) and glucocorticoids for induction of remission are accepted as a standard treatment for proliferative LN, and the standard monthly dose of 0.5 to 1 g/m2 provides an acceptable balance of the duration needed to achieve renal remission with the risk of gonadal and other toxicities.13 Nonetheless, cyclophosphamide (CY) can lead to severe adverse effects, such as infection, malignancies, and ovarian failure.4,14–16 Thus, another immunosuppressive drug, such as mycophenolate mofetil, tacrolimus, or mizoribine (MZR), may provide high efficacy and also have a better safety profile.
MZR was originally used as an antibiotic against Candida albicans, but researchers found it had strong immunosuppressive activity in various experimental animal models.17 MZR is an imidazole nucleoside and its metabolite, MZ-5-P, inhibits inosine monophosphate (IMP) synthetase and guanosine monophosphate synthetase, thereby completely inhibiting guanine nucleotide synthesis and halting DNA synthesis. An immunosuppressant, MZR has selective inhibitory effects on inosine 5-monophosphate dehydrogenase (IMP-DH), a key enzyme in the de novo synthesis of purine nucleotides. This suppresses the proliferation of T lymphocytes and B lymphocyte, and manifests as inhibition of cell-mediated and humoral immune responses.18–21
The combination of glucocorticoids with IVCY is an effective treatment commonly used for patients with LN in China, but this treatment regimen has some disadvantages. For example, multi-target therapy using mycophenolate mofetil and tacrolimus is more effective than IVCY for inducing complete remission in patients with advanced LN (class V or IV22), for which glucocorticoid + IVCY is considered unsatisfactory. Kagawa et al. previously reported that MZR and tacrolimus with corticosteroids were well tolerated and provided an effective alternative multi-target therapy that can induce remission in patients with LN.23 The proposed study will compare the efficacy and safety of MZR treatment with standard IVCY treatment for patients with LN.
A total of 250 participants from 44 hospitals in China were enrolled. A list of study sites is available as Extended data.24 We followed the SPIRIT checklist for reporting the protocol.24
The inclusion criteria are: diagnosis with SLE according to 1997 American College of Rheumatology criteria; kidney biopsy within 365 days prior to screening and confirmation of LN class III, III+V, IV, IV+V, or V according to the 2003 pathologic classification of the International Society of Nephrology/Renal Pathology Society; 24 h urinary protein level of 1.0 g or more; SLE disease activity index (DAI) of eight or more; male or female and between 18 and 70 years old (inclusive) at the time of providing informed consent; body weight between 40 and 80 kg (inclusive) at screening; and provision of written informed consent.
The exclusion criteria are: history of allergy to an investigational agent (MZR or CY) or glucocorticoids; accumulated CY dose of more than 3 g during the one year prior to screening; use of an immunosuppressant or a Chinese traditional medicine with an immunosuppressive effect within 30 days prior to screening; use of more than 1.0 mg/kg/day of prednisone or the equivalent dose of another oral glucocorticoid within 30 days prior to screening; use of other investigational drugs within 30 days prior to screening; use of plasma exchange therapy or immunoadsorption therapy within 30 days prior to screening; requirement for pentostatin or a live vaccine (not including the flu vaccine); undergoing renal replacement therapy; receipt of a kidney transplantation; a malignancy; severe hypertension (systolic blood pressure > 160 mmHg or diastolic blood pressure above > 100 mmHg) which is not effectively controlled; white blood cell count of 3×109/L or less; serum creatinine (SCr) level of 176.8 μmol/L or more; level of aspartate transaminase (AST) or alanine transaminase (ALT) more than 3-times of the upper limit of normal; positivity for hepatitis B, hepatitis C, or HIV; other suspected infection according to chest computed tomography (CT) or laboratory test results; unsuitability for participating in the study in the opinion of investigators (due to uncontrolled diabetes, central nervous system lupus, lupus encephalopathy, active psychosis, osteonecrosis of the femoral head, fulminant hepatitis, peptic ulcer, etc.); pregnancy, currently breast feeding, or willing to become pregnant; and any other disease that would affect the evaluation of drug efficacy or safety.
If patients need prohibited medication, they are to be withdrawn from the study. Subject premature withdrawal criteria are as follows: 1) Withdrawal by subject: a subject decides to withdraw from the study (including both screening period and treatment period. 2) Screen failure: A subject does not satisfy the inclusion criteria or satisfy exclusion criteria at Visit 0 or Visit 1. 3) Protocol violation: any serious violation against inclusion or exclusion criteria found after the first dose of the investigational drug; A subject takes prohibited medications or prohibited therapy before the last planned visit. Other major violation. 4) Pregnancy: A subject becomes pregnant. 5) Subject’s poor compliance with study drug: subject’s poor compliance with the protocol, including refusal of continued treatment or observation. 6) Investigator’s decision: any medical condition, including those listed under the exclusion criteria of the protocol, or personal circumstances, which in the opinion of the investigator, exposes the subject to substantial risk by continuing in the study or does not allow the subject to adhere to the requirements of the study protocol. 7) Adverse event: the occurrence of any clinically significant adverse event or serious adverse event, which in the opinion of the investigator warrants the subject’s premature withdrawal. If an adverse event is considered by the investigator as not clinically related to the study drug or if the benefits of continued study treatment are considered to outweigh the importance of the adverse event, the treatment may be continued at the discretion of the investigator. 8) Lost to follow-up: A subject does not come to a site for any reason before the planned last visit or the end of study visit. 9) Achieving endpoint event (progression to End-Stage Renal Disease” or “Doubling of serum creatinine (SCr)” through the study). 10) Others: it’s difficult for the subject to continue the study judged by the principal investigator or co-investigators.
The study is a multi-center, open-label, randomized controlled trial, which consists of a one-week screening phase followed by a 52-week treatment phase (Figure 1). For all eligible subjects, there will be a seven-day run-in screening period (day −7 to day −1, visit 0 [V0]) with no changes in the ongoing corticoid prescription. At day one (V1), subjects will be randomized into an MZR group or IVCY group in a 1:1 ratio stratified by renal biopsy pathology classification using permuted blocks by an Interactive Web Response System (IWRS).
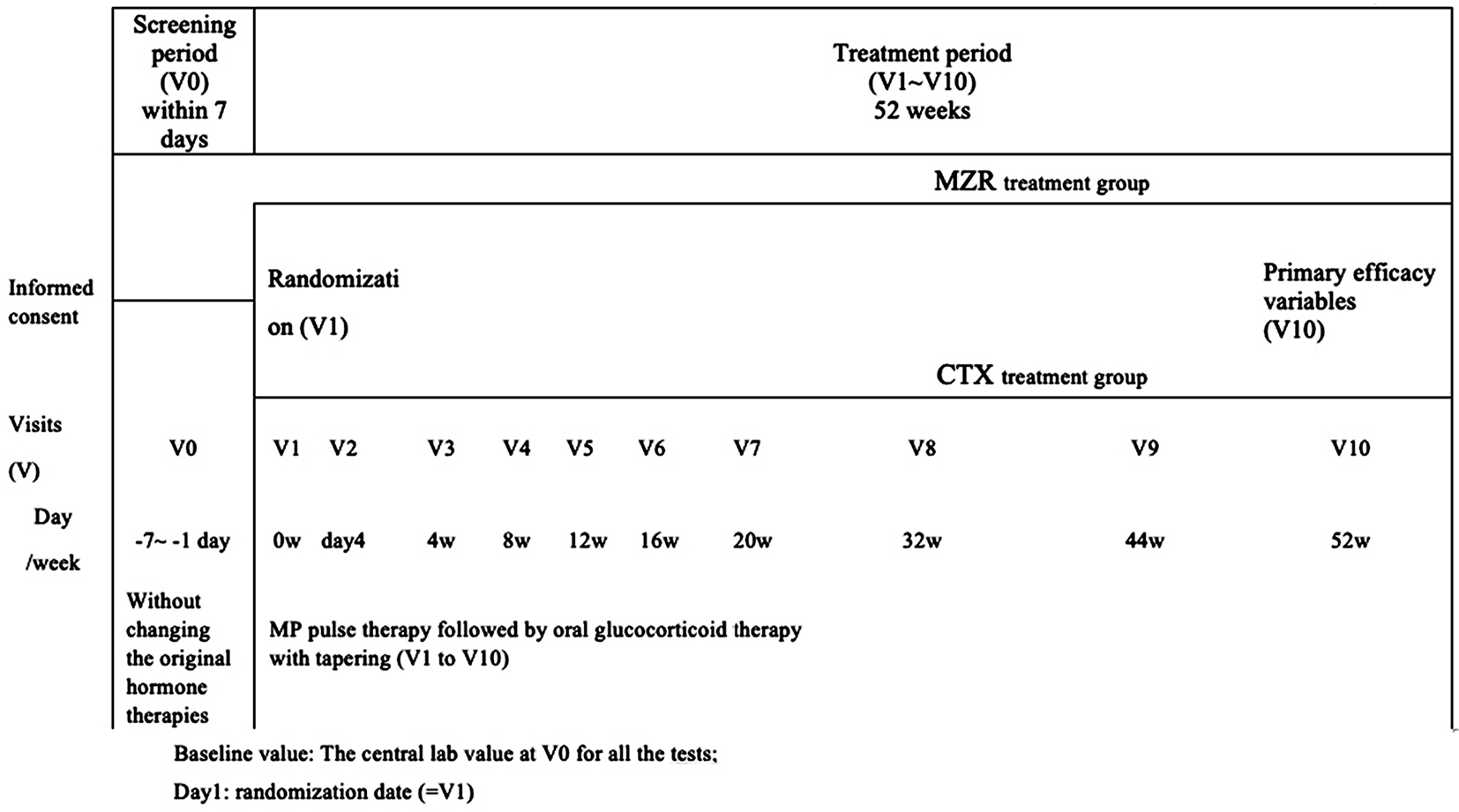
Medidata Balance is used as the IWRS system in this study. All required information was collected to complete the “Balance Design and Rave Integration Specification”, this file was sent to Medidata for Balance setup (Randomization Part). Once all settings pass Medidata internal quality control, the random number is generated. All settings will be copied to the production environment by Medidata and will be ready for go-live. The number of pathological types in the random stratification factors are three, and the pathological types are [III, III+V], [IV, IV+V], and [V only]. The site is not stratification factor. The random sequence for allocation (i.e. computer-generated random numbers) will be generated in the IWRS. When an investigator enrolls an eligible patient in the IWRS, a random sequence for allocation will be displayed on it, and the investigator will assign the participant to the intervention based on the information from the IWRS.
During the 52-week treatment period, the subjects will be required to return to the study center every four weeks (V2 to V7), then every 12 weeks (V8, V9), and then after eight weeks (V10) for assessments of each parameter and for collection of blood and urine samples (Figure 2, Table 1).
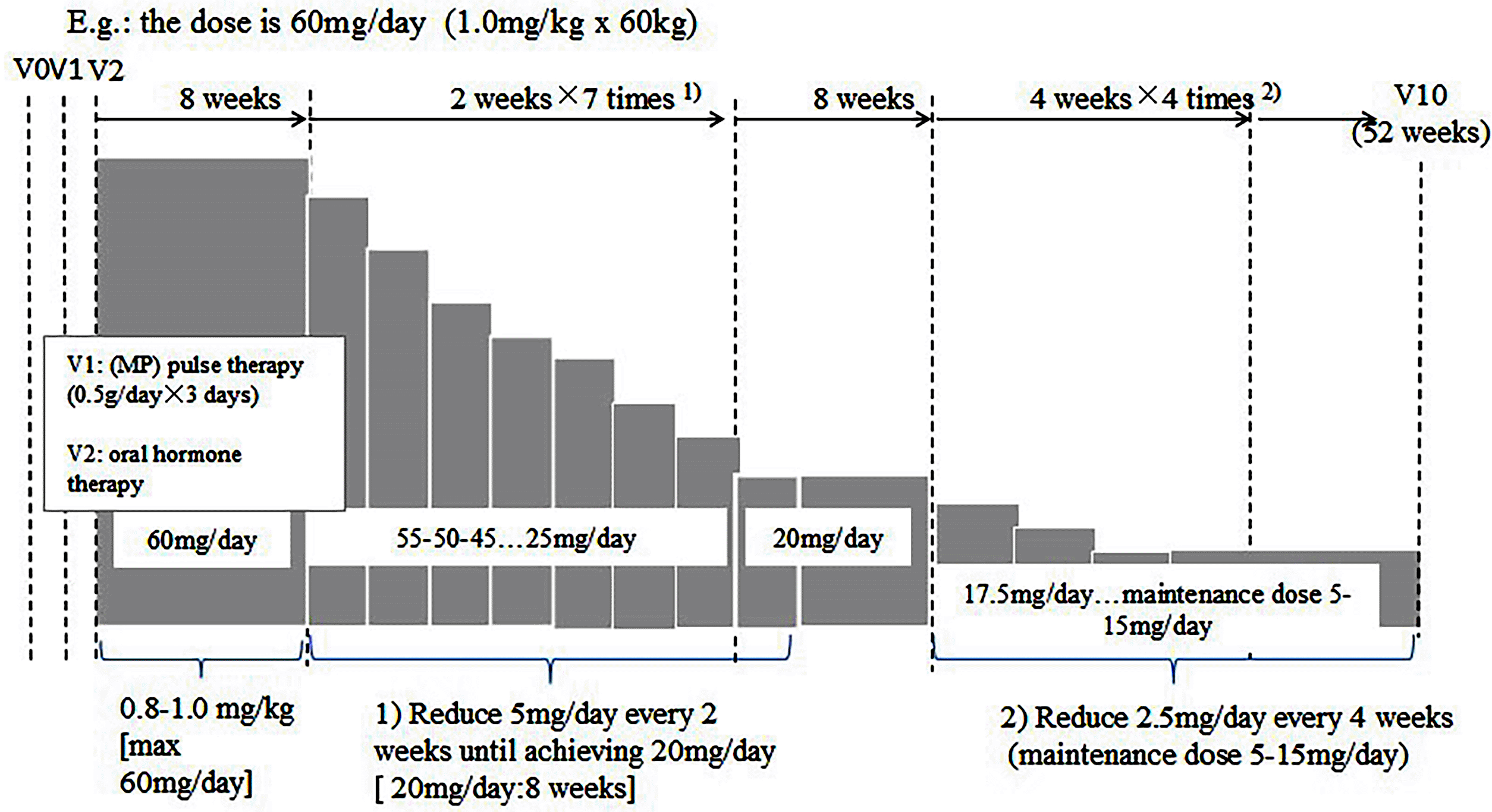
For subjects randomized to the MZR group, methylprednisolone (MP) pulse therapy (0.5 g/day for three days) will begin on V1, and oral corticoid treatment will then be started following standard guidelines (see below). Starting from V2, 150 mg per day of oral MZR will be given (one 50 mg/tablet t.i.d.).
The standard oral corticoid therapy guidelines for V2 to V10 are oral corticoid therapy (0.8–1.0 mg/kg/day prednisone or equivalent dose of MP) for eight weeks (V2 to V4). The daily oral steroid dosage will not exceed 60 mg/day, and subjects weighing 80 kg will be given 60 mg/day. After eight weeks (V4), steroid-tapering will begin. The dose reduction will be 5 mg/day every two weeks until the dose is 20 mg/day. Dosing with 20 mg/day will continue for eight weeks. After that, dose reduction will be 2.5 mg/day every four weeks. The dose will be maintained between 5 and 15 mg/day up to week 52 (V10) (Table 2).
For subjects randomized to the IVCY group, the MP pulse therapy and oral steroid treatment will be the same as in the MZR group. Each IVCY dose will be given at V2 to V7 (every four weeks) and at V8 and V9 (every 12 weeks). The dosage will be calculated based on body surface area (BSA, m2) and will range from 0.5 g/m2 (minimum) to 1.0 g/m2 (maximum). BSA will be calculated using the Dubois & Dubois formula:
When the subject’s condition faces deterioration eight weeks after randomization (V4), the original dosage of hormone therapies (at most) can be maintained for four weeks. After the end of adjusted hormone treatment, the investigator will assess the subject’s condition again. If the subject’s condition still meets the definition of deterioration, the subject should withdraw from the study.
Deterioration is defined as 24 hours urine protein level greater than 150% of the baseline value or worsening Systemic Lupus Erythematosus Disease Activity Index (SLE-DAI) score in comparison with baseline.
For example, if the subject’s condition deteriorates at Visit 4, the dose of hormone therapies should be continued for two to four weeks (Table 2).
The primary efficacy variable will be total remission (TR, %) after the entire treatment period (V10), defined as the sum of complete remission (CR, %) and partial remission (PR, %) at that time. CR will be defined by the presence of all of the following criteria: 24 h urine protein below 0.3 g; serum albumin 35 g/L or more; and SCr level within the normal range, or not more than 25% above the baseline (V0) or decreased relative to baseline (V0). PR will be defined by the presence of all of the following criteria: 24 h urine protein decreased by 50% or more from the baseline (V0) and below 3.5 g; serum albumin 30 g/L or more; SCr level not more than 25% above the baseline (V0) or lower than the baseline; and lack of CR.
The secondary efficacy variables will be complete remission rate and partial remission rate; treatment failure (i.e., not achieving at least partial remission); 24 h urine protein and serum albumin; SCr, estimated glomerular filtration rate (eGFR) determined using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula, and blood urea nitrogen (BUN); C3, anti-DNA antibodies, anti-nuclear antibodies, anti-Sm antibodies, and anti-phospholipid antibodies; SLEDAI score; and any endpoint event during the study, defined as progression to ESRD (need for long-term dialysis or renal transplantation) or doubling of SCr relative to baseline.
Safety will be assessed by measuring the following variables: any reported adverse event, including severe adverse events, pregnancy, important treatment-related adverse events (granulocytopenia, infection, hemorrhagic cystitis, liver function impairment, hyperuricemia, malignancy, amenorrhoea, alopecia, nausea/vomiting), and adverse events leading to study discontinuation; laboratory test results, including routine blood cells tests, blood biochemistry, routine urine tests, and IgG; body weight; vital signs, including blood pressure, pulse, and body temperature; 12-lead electrocardiogram in the resting state; and chest CT.
The routine blood cell test will measure hemoglobin, hematocrit, white blood cells (WBCs), platelets, and red blood cells (RBCs). The blood biochemistry test will measure ALT, AST, total bilirubin, total protein, albumin, uric acid, total cholesterol, triglycerides, glucose, SCr, eGFR using the CKD-EPI formula,25 and BUN. The urine (dipstick) test will measure urine protein and glucose (at V0, V7, and V10), urine type, RBCs per high-power field (HPF), and WBCs/HPF.
Laboratory tests will be performed by a central laboratory. The investigator will evaluate the clinical significance of each laboratory value outside the reference range. When the results from a local laboratory differ from those of the central laboratory, those of central laboratory will be used.
The time schedule of enrollment, interventions, assessments, and visits for participants are shown in Table 1 and summarized in Figure 1. The first study site was visited in November 2014, with the first participant enrolling in November 2014. The last participant was enrolled in March 2018 and the study was completed in March 2019.
The investigator recruited subjects from patients who attended the study center and placed ethically approved recruitment advertisements at the study center. If necessary, the investigator commissioned recruitment centers to recruit subjects in accordance with the principles of recruitment and retention in clinical trials.
Non-inferiority testing will be used to assess the primary efficacy variable, as the relative risk ratio of TR (%) in the two groups after 12 months (52 weeks) of treatment for the per-protocol set (PPS) population. The sample size of 125 patients per group (total 250 patients) was determined based on a TR of 73% for both groups,26,27 a non-inferiority difference of 0.726, a power of 90%, a one-sided significance level of 2.5%, and a drop-out rate of 20%.
All data processing, summarization and analyses will be performed using Version 9.1.3 (or later) of the SAS® statistical software package (Windows OS). Relative risk ratio (MZR versus CTX) and its 95% confidence intervals (two-sided at α-level of 5%) will be calculated and non-inferiority testing will be applied. If the lower limit of the relative risk ratio for the PPS population is greater than 0.726, then the non-inferiority of MZR and CTX can be claimed.
The full analysis set (FAS) will include all subjects who were randomized and received at least one dose of a study drug and at least one post-treatment efficacy assessment. The PPS will include all FAS subjects without any other major protocol violation. The safety set (SS) will include all randomized subjects who received at least one dose of a study drug and had at least one subsequent safety assessment. Safety analysis will be based on the actually assigned treatments.
The primary efficacy variable is TR (%) after 12 months of treatment. A noninferiority test of TR (%) will be performed in the PPS population by comparing the two groups at V10. The relative risk ratios and two-sided 95% confidence intervals will be calculated.
The secondary analysis for the primary efficacy variable will be performed in the PPS at the end of the study and in the FAS at V10. The number of subjects who achieved TR will be calculated in each group at each visit in the PPS population. The secondary efficacy variables will be analyzed using descriptive statistics.
Subgroup analysis will be performed as necessary. The following subgroup analyses are planned: pathologic classification, eGFR, C3, anti-DNA antibodies. Additional subgroup analyses will be pre-specified in the statistical analysis plan before locking the database. All safety data will be analyzed descriptively. High sensitivity C reactive protein (hs-CRP) is an exploratory variable that will be measured during the study visits V0 and V3 to V10, and values will be summarized at V4, V7, V8, V9, and V10.
A data monitoring committee was not set up in this study because MZR has a high safety profile and was approved as an indication for LN in Japan as early as 1990. The primary endpoint of this study is to compare the overall response rate between the MZR and CTX groups after 52 weeks of treatment, therefore no interim analysis is planned. However, the criteria for suspension of investigational drug use and the subject’s premature withdrawal are clearly defined in the protocol.
The CRO’s QA department and authorized third-party auditors have undertaken monthly quality audits of the clinical trial. All medical records, the trial related files and correspondence, as well as the informed consent document, are accessible to the quality auditor.
This study was approved by the Medical Ethics Committee of the Chinese People’s Liberation Army General Hospital on 15 September 2014 (C2014-041-01). Written informed consent for publication was obtained from all participants. The trial was registered on ClinicalTrials.gov: NCT02256150 (Registered 2014-10-01).
Participant data is pseudonymised by assigning each participant an identifier number, which is used to identify the participant during the study and for any participant specific communication between the investigator and sponsor. These enrollment lists must be kept strictly confidential by the investigator and cannot be shared with the sponsor.
The investigator or authorized site staff will enter data into the eCRFs. Only medically qualified (sub)investigators are permitted to sign clinical assessment/safety data. Any changes made to the eCRF by the investigator or authorized site staff after the original entry will be automatically recorded in the system.
According to the protocol, whether subjects complete the study visit or withdraw prematurely from the study visit, they must be assessed for all indicators at the last visit. Following the completion of the clinical trial, the investigator should monitor all adverse events and submit a follow-up report to the sponsor until they resolve, stabilize, the patient is lost to follow-up, or alternative explanations for the event can be provided (In the case of permanent damage, follow up until the adverse event is considered stable). Female subjects must notify the investigator and withdraw from the study if they become pregnant. The subject should be informed of the risks of continuing the pregnancy by the investigator and be monitored until the end of the pregnancy.
For this trial, the sponsor purchased “Human Clinical Trial” insurance from Chubb Insurance Company Limited and has included the following provision in the informed consent form for indemnification for risks resulting from involvement in the trial: From the enrollment in the trial to the end (duration 52 weeks), if any adverse event occurs, the investigator must determine whether it is related to the investigational drug and the diagnostic tests required by the protocol. If so, the participant can take treatment at the hospital where she/he participated in the trial, and the sponsor will be responsible for all medical expenses and financial compensation as required by law. However, the sponsor will not cover any costs that are unrelated to this trial. If the participant files a claim for compensation following any adverse event, the sponsor will contact the insurance company, then the insurance company will pay the compensation in accordance with the insurance contract.
The current study aims to evaluate the efficacy and safety of oral MZR in comparison with standard IVCY for treatment of LN. The efficacy endpoints are CR (%), PR (%), and treatment failure; 24 h urine protein and serum albumin; SCr, eGFR, and BUN; C3, anti-DNA antibody, ANA, anti-Sm antibody, and anti-phospholipid antibody; SLE-DAI; and attainment of any endpoint event during the study period. The exploratory objective of this study is to evaluate changes of hs-CRP in subjects who receive oral MZR compared with IVCY for treatment of lupus nephritis.
This study is the largest RCT to examine MZR, and also has the longest intervention and observation times among all large RCTs that examined MZR. Previous studies showed that despite aggressive treatment, the proportion of patients with renal responses was very low, and about 60% of patients did not achieve complete remission.28 Moreover, 27 to 66% of LN patients in remission experienced relapses,29 an important factor when considering exacerbations of renal damage and poor prognosis.30 Therefore, this study is designed to provide more therapeutic options and effective maintenance therapy for LN by studying the effects of MZR using a larger sample size and a longer observation period.
This study had an open label design rather than a double-blind design because of the different dosages and routes of administration of the study drugs. In particular, for a double-blind design all subjects would be required to receive oral MZR or oral placebo three times per day and eight intravenous injections of CYC or intravenous placebo, which would clearly be a significant treatment burden. In addition, LN can be dangerous in some cases, and use of double-blinding when serious adverse events occur may delay the timing of treatment. Therefore, an open study design will be used to ensure the safety of all subjects and reduce the burden of treatment.
The clinical efficacy of MZR as an immunosuppressant for renal transplantation was investigated in several Japanese institutions from 1978 to 1982 and MZR was first approved by the Japanese Ministry of Health, Labor and Welfare (MHLW) for the prevention of rejection following renal transplantation in 1984.31 The MHLW also approved MZR for the treatment of LN in 1990, rheumatoid arthritis in 1992, and primary nephrotic syndrome in 1995.31 In all these diseases, MZR is often used in combination with corticosteroids and/or anti-inflammatory drugs.
Previous clinical trials and post-marketing surveillance studies examined more than 4000 patients who received MZR for kidney transplantation, LN, rheumatoid arthritis, and nephrotic syndrome. The results of these studies showed that MZR was well-tolerated and had a good safety profile. MZR was approved in China for suppression of rejection following renal transplantation in 1999.
This paper describes the protocol of a multi-center, open-label, randomized controlled trial that compares the efficacy and safety of MZR with IVCY for treatment of LN. This study is the largest RCT to examine MZR, and also has the longest intervention and observation times among all large RCTs that examined MZR. The proposed study will examine the suitability of an additional indication for this drug in China.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)