Keywords
leishmaniasis, Leishmania arginase, computer-aided drug design, molecular dynamics simulation, antiprotozoal agents; drug discovery
This article is included in the Cheminformatics gateway.
leishmaniasis, Leishmania arginase, computer-aided drug design, molecular dynamics simulation, antiprotozoal agents; drug discovery
We have modified the current version of the manuscript attending the 2nd reviewer's suggestions. We have added information regarding the identity of the parasite and human targets at the results section.
See the authors' detailed response to the review by Francisco Centeno
See the authors' detailed response to the review by Anupam Nath Jha
Leishmaniasis is an ancient disease that has been described in archaic ceramics, statues, and writings, and in molecular findings from mummified human bodies and archaeological material.1 The disease causes high morbidity and mortality worldwide, where about one billion people are at risk of infection across 98 countries, with over 1.5 million new cases and 20,000-40,000 deaths reported each year.2,3 The increase in leishmaniasis incidence and prevalence is mainly attributed to several risk factors that are man-propelled,4 whereas, in many regions, the transmission pattern shows expansion, with new territories affected by the disease.5,6 Also, leishmaniasis has gained greater importance in HIV-infected patients as an opportunistic infection in areas where both pathogens are endemic.7 Leishmaniasis is caused by the protozoan parasites of the genus Leishmania (Kinetoplastida: Trypanosomatidae), which has a digenetic life cycle that alternates between the midgut of sandflies and the phagolysosomes of mammalian macrophages.8 When exposed to extreme environmental changes, such as low pH, the parasites respond to the acidification of their environment by changing the pattern of expression of several proteins.9,10 About 21 parasite species can infect mammals and many of them cause human disease11 and the clinical manifestations depend on both the parasite species and the hosts’ immune response,12 varying from a chronic, slow-to-heal disease known as tegumentary leishmaniasis (TL), to a potentially fatal form of the disease, namely, visceral leishmaniasis (VL), in which parasites disseminate to internal organs, such as the liver, spleen, and bone marrow.13
Despite significant progress, the development of a human vaccine remains hampered by significant gaps in the development pipeline14; and the treatment against disease has used drugs that cause side effects in the patients, such as myalgia, arthralgia, anorexia, fever, and urticaria, as well as toxicity in the liver, kidneys, and spleen.15 Therefore, the necessity for cost-effective treatment which promotes the cure completely, with few side effects, low relapse rates, high effectiveness, and a reduction of toxicity remains.16 The number of drugs derived from natural products (NPs) present in the total amount of drug launchings in the market over four decades represents a significant source of new pharmacological entities,17 while a series of secondary plant-purified products has already been described with leishmanicidal potential.18–21 Likewise, computer-aided drug design (CADD) can be defined as computational approaches that are used to discover, develop, and analyze drug and active molecules with similar biochemical properties,22 and this has become crucial for screening of potential metabolite databases from natural sources that can be repurposed against diseases for faster, safer, and cheaper drug development.23,24 The strategy of target-based drug discovery is used extensively by the pharmaceutical industry and has been applied to leishmaniasis.25,26 However, in silico methods to identify new potential drugs to be applied against leishmaniasis present limitations, such as the dependency on the quality, accuracy, and completeness of the information present in databases.27 The arginase (ARG) enzyme has recently obtained considerable attention since new studies have highlighted it as a potential therapeutic target in leishmaniasis.28 ARG is the first enzyme of the polyamine pathway and catalyzes the conversion of L-arginine to L-ornithine and urea, down-regulating the polyamine pathway, affecting the parasite growth and infectivity.29 The inhibition results in a lack of protection against reactive oxygen species (ROS), which damages Leishmania’s genetic material and ultimately leads it to die by apoptosis.30 As a result, various NPs have demonstrated anti-arginase action,31,32 and the majority of these NPs have also demonstrated a strong affinity against human ARG.33 In the current study, we used CADD techniques, such as virtual screening, molecular docking, and molecular dynamics simulations, to identify structural analogs of NPs that have demonstrated anti-leishmanial and anti-ARG activities and that may bind specifically to the Leishmania ARG. Our goal was to identify a promising compound candidate that could be used in the treatment of leishmaniasis.
The search for natural products with anti-leishmanial and anti-ARG activities was performed at the Nuclei of Bioassays, Ecophysiology, and Biosynthesis of Natural Products Database (NuBBEDB) online web server (version 2017) (http://nubbe.iq.unesp.br/portal/nubbe-search.html, accessed on 23 January 2022), which contains the information of more than 2,000 natural products and derivatives34; while the “anti-leishmanial property” was selected in the biological properties segment of the web server. The bibliographic data extraction, regarding the compounds found in NuBBEDB, was performed from the National Center for Biotechnology Information (NCBI) databases (https://www.ncbi.nlm.nih.gov/pubmed/, accessed on 07 February 2022); and the simplified molecular-input line-entry system (SMILES) was searched and retrieved from PubChem server (https://pubchem.ncbi.nlm.nih.gov/, accessed on 10 February 2022).35 Likewise, the physicochemical properties: total molecular weight (MW), octanol/water partition coefficient (iLOGP), number of H-bond acceptors (HBAs), number of H-bond donors (HBDs), and the topological polar surface area (TPSA), for each compound were calculated within the Osiris DataWarrior v5.2.01 software36; and, the rotatable bonds (RB); number of heavy atoms (NHA); and synthetic accessibility (SynAcce) were calculated within SwissADME server (http://www.swissadme.ch/index.php, accessed on 15 February 2022).37
The SMILES from the compounds were used for high throughput screening to investigate structural analogs by the SwissSimilarity server (http://www.swisssimilarity.ch/index.php, accessed on 01 March 2022)38; whereas the commercial class of compounds was selected and the Zinc-drug like compound library, which comprises 9’205’113 molecules, with the combined screening method, was chosen for the high throughput screening to achieve the best structural analogs. The zinc-drug like compound library selection allowed the screening of compounds in the subsequent commercially available chemical libraries: Enamine, ChemBridge, Maybridge, Asinex, AsisChem, Otava, SPECS, TimTec, Vitas, Life Chemicals, ChemDiv, and Innovapharm.39 Threshold values for positivity were selected by default parameters. Also, the FASTA sequences of the ARG sequences from L. infantum (A4IB49), L. mexicana (Q6TUJ5), L. braziliensis (A4HMH0), and Homo sapiens (P05089) were retrieved from UniProt database (http://www.uniprot.org/, accessed on 03 March 2022) (RRID:SCR_002380), and subjected to automated modeling in SWISS-MODEL40 (RRID:SCR_018123), whereas the best model was selected based on the GQME and QMEAN4 scores.
Furthermore, the compounds were imported into Open Babel (RRID:SCR_014920) within the Python Prescription Virtual Screening Tool41 and subjected to energy minimization. PyRx (RRID:SCR_018548) performs structure-based virtual screening applying molecular docking simulations using the AutoDock Vina tool42 (RRID:SC_011958), whereas the drug targets were uploaded as macromolecules. For the analysis, the search space encompassed the whole of the modeled 3D models, and the molecular docking simulation was then run at an exhaustiveness of 8 and set to only output the lowest energy pose. The Osiris Data Warrior software was employed to calculate the potential tumorigenic, mutagenic, and reproductive effects, and irritant action of selected compounds predicted by comparison with a precompiled fragment library derived from the Registry of Toxic Effects of Chemical Substances (RTECS) database.36
Ligands preparation was based on the results from the virtual screening analysis; while the geometry optimization of these compounds was made in the Avogadro v. 1.2.0 program43 (RRID:SCR_015983) and the ACPYPE (AnteChamber PYthon Parser interfacE)44 server was employed to generate the topologies and parameters for molecular dynamics (MD) simulation. We determined the 3D structural conformation of L. infantum ARG by homology modeling with L. mexicana ARG (PDB ID: 4ITY) as a template in the SWISS-MODEL online server40 and afterwards we determined the protonation/deprotonation states at pH 2.0 and pH 7.0 in the PDB2PQR.45 Since ARG is a trimeric metalloprotein with three active sites binding to two manganese atoms (Mn+2), we fixed the Mn+2 coordination with active site residues and a hydroxyl molecule (OH−1), considering the following coordination: first Mn+2 with His114 (ND1), ASP137 (OD2), ASP141 (OD2), ASP243 (OD2) and the second Mn+2 with ASP137 (OD1), HIS139 (ND1), ASP243 (OD1) and ASP245 (OD2). The MD simulation was reproduced in GROMACS v. 202046 (RRID:SCR_014565), considering the AMBER9947 force field. The systems were solvated with the TIP3P water model, and Na+1 or Cl−1 ions were added for neutralization. The box size was 12×12×12 nm. Thus, the energy minimization was performed with the steep-descent algorithm with 20000 steps of calculation. The MD simulation was done in two steps; the first step was in the canonical ensemble (NVT) considering distance restraint of Mn+2 to the active site by 5 ns. The second step was the MD production in the isothermal-isobaric ensemble (NPT) with a time of 100 ns. The V-rescale48 thermostat was used to regulate the temperature at 309.65 K and the Parrinello-Rahman barostat at a reference pressure of 1 bar. Molecular docking was done with the DockThor online server49; in the last frame, the molecular docking at two pH conditions was used as a receptor. A grid was considered in the active site of ARG (ChainA). The complex models with the best scores were chosen, and these were subsequently simulated in the isothermal-isobaric ensemble NPT for 100 ns. Gibbs free energy was calculated by the molecular mechanics-generalized Born surface area (MM-GBSA)50 method in gmx_MMPBSA tool based on AMBER’s MMPBSA.py, and AmberTools2051 (RRID:SCR_014565) package was used. Additionally, to compare the binding free energy studies, we include the free energy perturbation (FEP) analysis where the Bennett acceptance ratio (BAR) calculates the free energy differences.52 This analysis is achieved with the free energy implementation by the GROMACS tool.
Results were entered into Microsoft Excel (version 10.0, Microsoft Corporation, Redmond, WA, USA) spreadsheets and analyzed by GraphPad Prism version 9.4.0 for Windows, GraphPad Software, San Diego, California USA, (http://www.graphpad.com) (RRID:SCR_002798). To evaluate the correlation between the binding affinities of the compounds against the protein targets, they were placed in a linear regression plot and analyzed by Pearson’s correlation coefficient; differences were considered significant when p<0.05. Further, the selectivity score of binding affinities was calculated as described53; where a selectivity value >1 indicates a priority of the compounds to bind to the parasite ARG over the human target. Heatmaps were constructed in the R programming environment (version 4.0.3) using the “heatmap 2” function in the package “gplots”.54
In this work, a search was performed in the NuBBEDB for NPs that had been described with anti-leishmanial and anti-ARG activities. The search in the database resulted in 33 NPs described with anti-leishmanial activity, whereas six of them had also been described as inhibitors of ARG activity. Startlingly, all the NPs selected were described in the same article, in which the compounds were isolated from Byrsonima coccolobifolia species and tested for in vitro anti-ARG activity.55 Since no anti-leishmanial activity was reported in the article, a cross-reference search for each compound was performed in the PubMed database to validate the properties. Thereafter, the SMILES from quercetin (NuBBE_122), isoquercetin (NuBBE_123), quercitrin (NuBBE_161), (+)-syringaresinol (NuBBE_214), catechin (NuBBE_287) and (-)-epicatechin (NuBBE_866) were obtained from PubChem and submitted to physicochemical properties analysis related to an absorption, distribution, metabolism, and excretion (ADME) profile; Lipinski’s rule of five (MW, iLOGP, HBAs and HBDs),56 the quantitative estimate of drug-likeness (TPSA, RB, NHA and the number of alerts for undesirable substructures)57 and the synthetic accessibility,58 of the NPs are shown in Table 1.
To find structural analogs to the six NPs selected, a search of the SwissSimilarity server employing the commercial zinc-drug like compound library was performed, resulting in 400 analogs for each NP; however, the search comprised a high degree of redundancy between the analogs and a step in which duplicated compounds were excluded was executed, resulting in a total of 1499 unique compounds selected for virtual screening (Figure 1). The virtual screening results against Leishmania infantum and human ARG, which shown a 44% of sequence identity, are plotted in Figure 1A, where a positive linear relationship between the binding affinities of the compounds toward both targets is shown [Pearson r:0.931; r2:0.868]. Later, aiming to select compounds that showed higher affinity toward L. infantum ARG, the selectivity was calculated, and compounds with scores >1 were screened, resulting in 25 compounds selected (Figure 1A). Since in vitro evidence of inter-species differences in the susceptibility of parasites to anti-leishmanial drugs has been reported,59 putative drug candidates must be active against several species of the parasite60; in this way, the selectivity of the compounds against L. mexicana and L. braziliensis ARG were also calculated and plotted in a heatmap; each compound’s results showed differences in their affinities profile (Figure 1B). Also, to select potential nontoxic candidates, the tumorigenic, mutagenic and reproductive effects, as well as irritant action were assessed for the 25 compounds (Figure 1C). Thus, the compounds 2H-1-benzopyran, 3,4-dihydro-2-(2-methylphenyl)- (9CI) (ZINC39120134) (Figure 1D), echioidinin (ZINC14807307) (Figure 1E), and malvidin (ZINC897714) (Figure 1F) were selected for further analysis, since they showed favorable binding affinities against the three parasite species targets and negative results for potential toxicities.
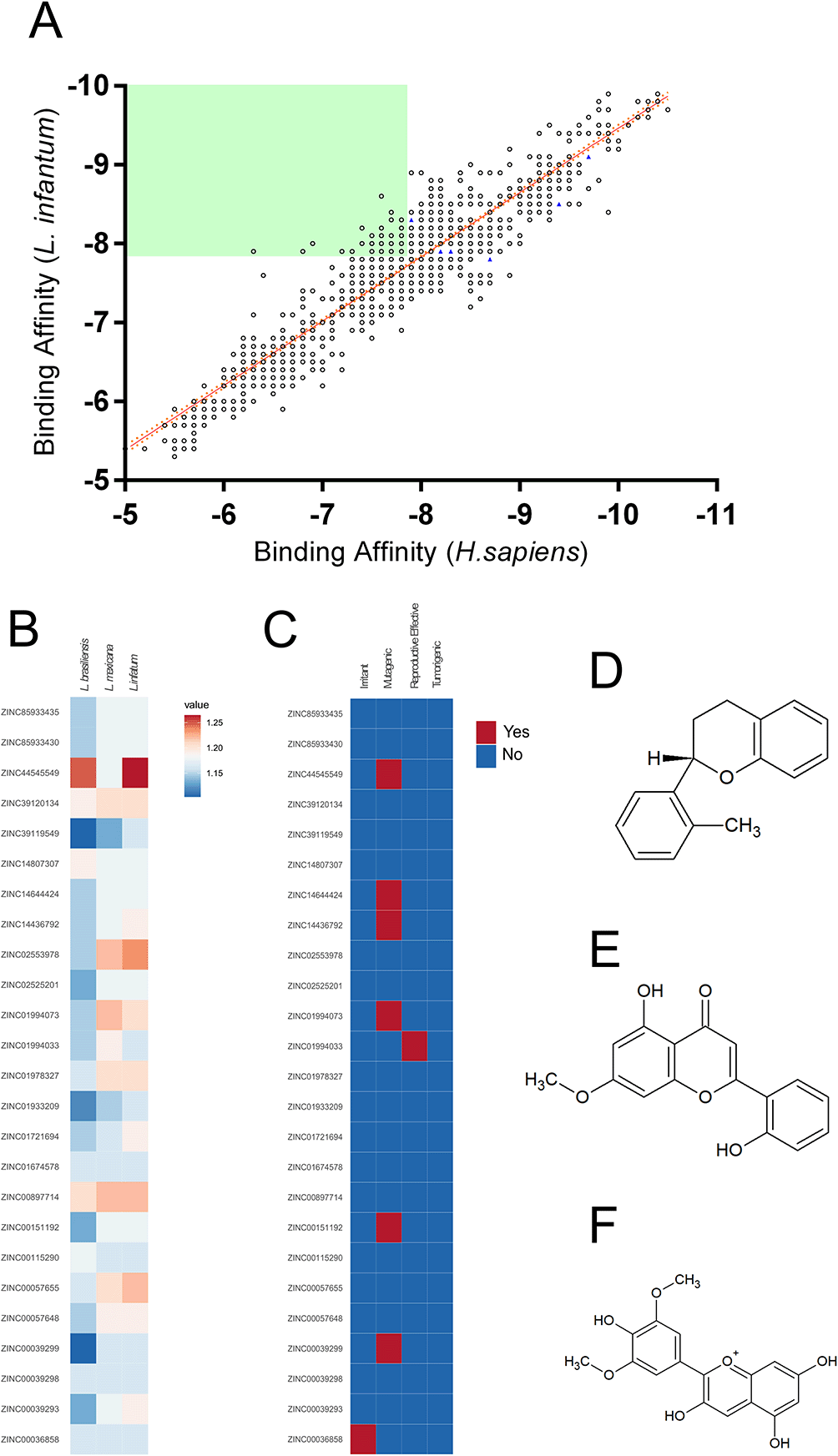
Binding affinities toward L. infatum and H. sapiens ARG targets were analyzed by linear regression and Pearson’s correlation coefficient. Solid orange line: linear regression; dotted orange lines: 95% confidence intervals. The solid green square was calculated using the maximum binding affinities of the 6 NPs (A). Normalized binding affinities heatmap of 25 selected compounds on L. infantum, L. mexicana, and L. braziliensis against their human homolog (B). Binary heatmap showing positive (red) or negative (blue) predicted toxicities (C). Chemical structure of ZINC39120134 (D), ZINC14807307 (E), and ZINC897714 (F).
L. infantum ARG is an enzyme with trimeric conformation (ChainA, ChainB, and ChainC) and its structure showed stable behavior during a 100 ns of MDS performed at pH 2.0 and pH 7.0 (Figure 2). Here we included the metal ions (Mn+2) and one hydroxyl molecule (OH−1) for each active site, and it was observed that some regions lose their structural conformation at pH 2.0 conditions (green color). In addition, compared to ARG at pH 7.0, ARG at pH 2.0 exhibits large structural alterations and high variations per residue (see Figure 3A and 3B). In Figure 3C, the radius of gyration shows lower compaction of whole protein during the MDS at pH 7.0 than at pH 2.0. The report of the trajectory of each complex system (enzyme-ligand) and the protein without ligand is shown in Figure 4. Since the root-mean-squared deviation (RMSD) is a noteworthy analysis to verify the similarity between a protein-bound and not bound ligand.61 The RMSD values in nm are presented that were taken from the ChainA of each protein in different pH conditions, whereas the enzyme-ligand systems presented greater conformational changes in the substrate-binding site (Figure 4A). Likewise, radius of gyration (RG) analysis verifies the compactness of protein structures, where the lowest RG demonstrates the tightest packing and high conformational stability.62 The results showed that, at pH 2.0, low compactness and a large broadening of the macromolecules are reported (Figure 4B). Figure 4C shows the root-mean-squared fluctuation (RMSF) per residue of the backbone, where high fluctuations were shown from residue 50 to 100 in both systems. From the enzyme-ligand simulation results, we take each simulation’s last frames (Figure 5). The compounds ZINC14807307 and ZINC897714 generate exciting interactions in the active center at the pH conditions evaluated and, at pH 7.0, hydrogen bonds are observed, which benefits enzyme-ligand coupling.
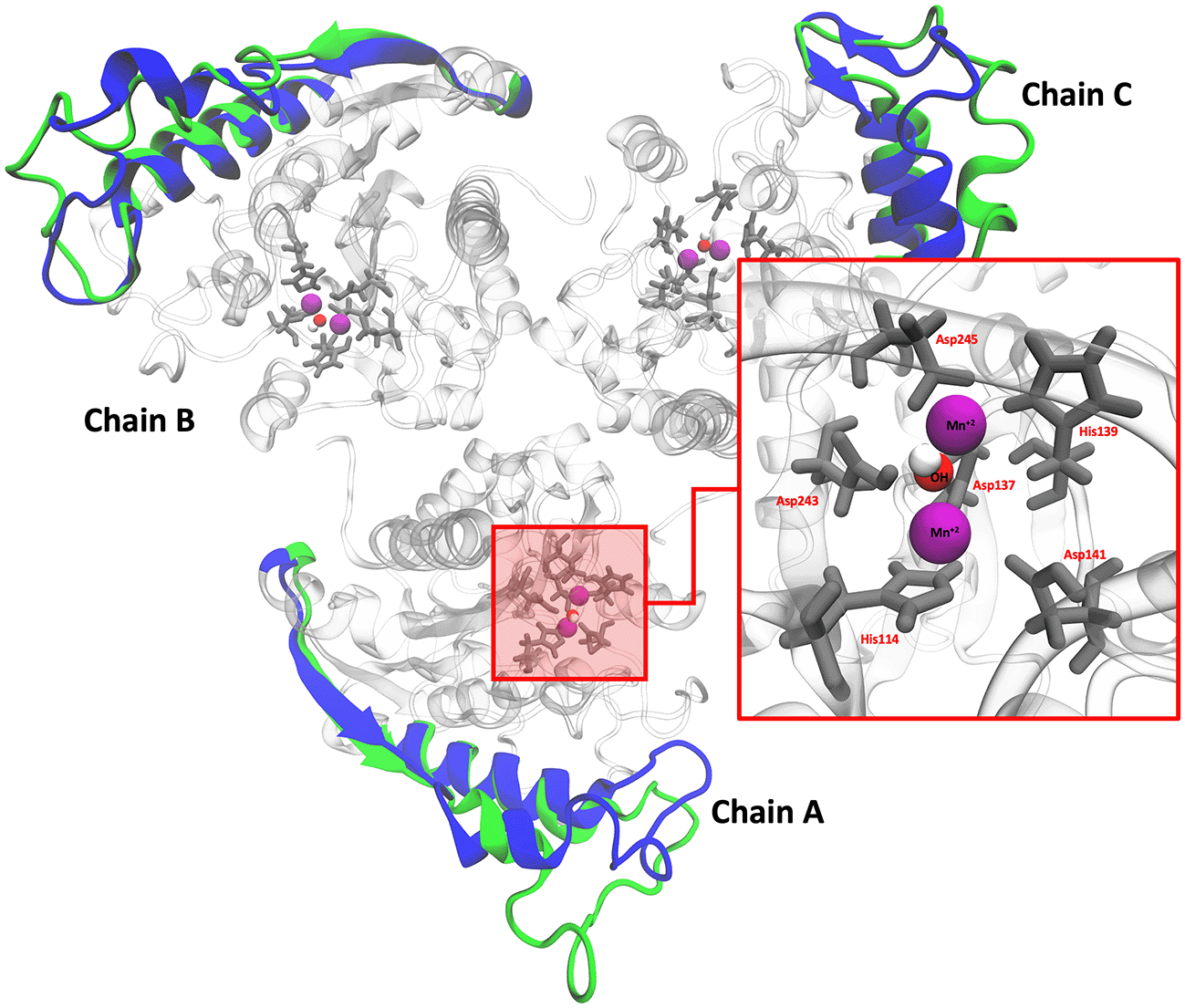
Colors blue and green represent the cartoon representation of pH 2.0 and pH 7.0. The red box shows the active site of ARG.
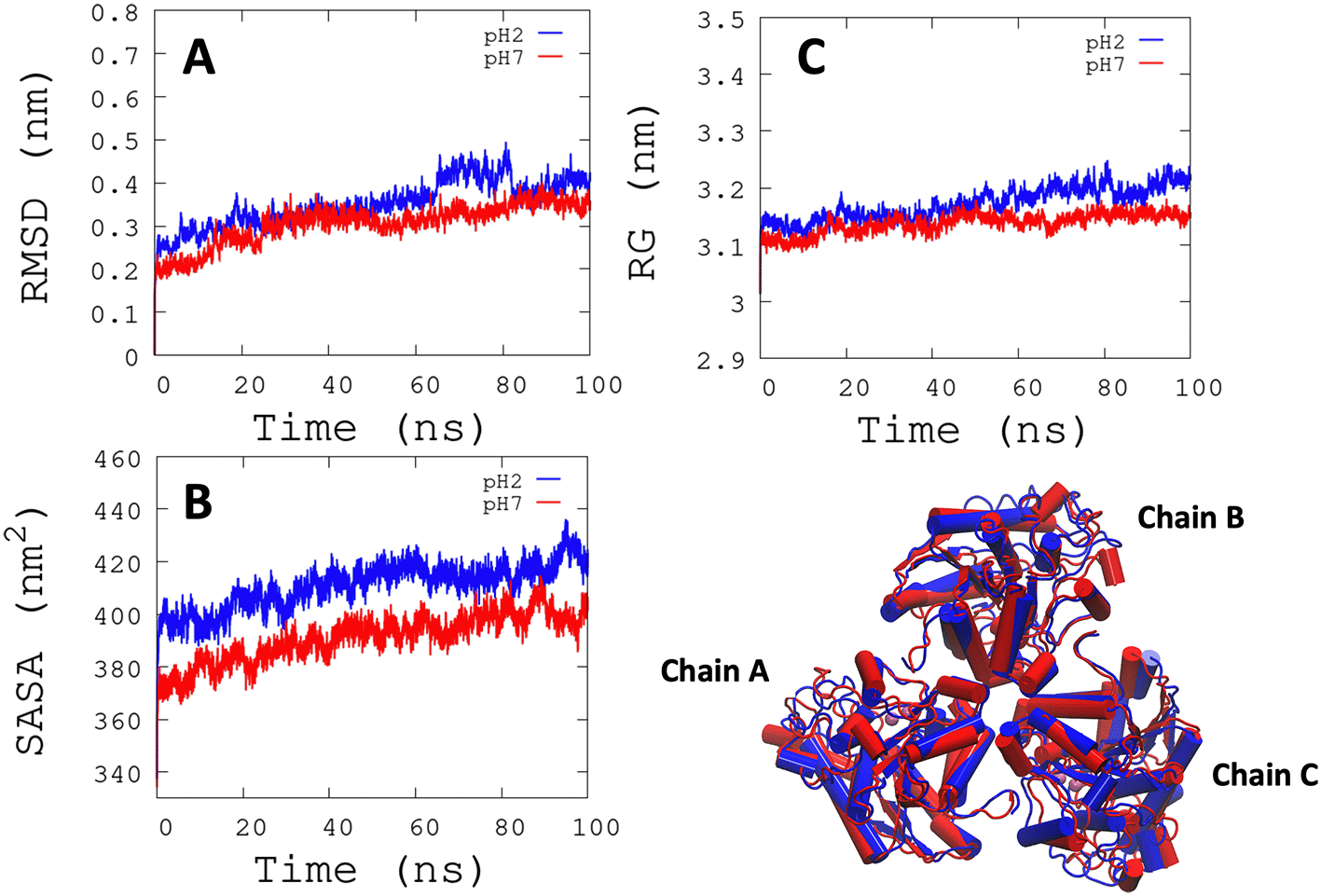
(A) RMSD is shown the conformational changes reported at pH 2.0. (B) SASA shows a greater solvent access surface area to ARG at pH 2.0 than at pH 7.0. (C) RG shows the same behavior as RMSD.
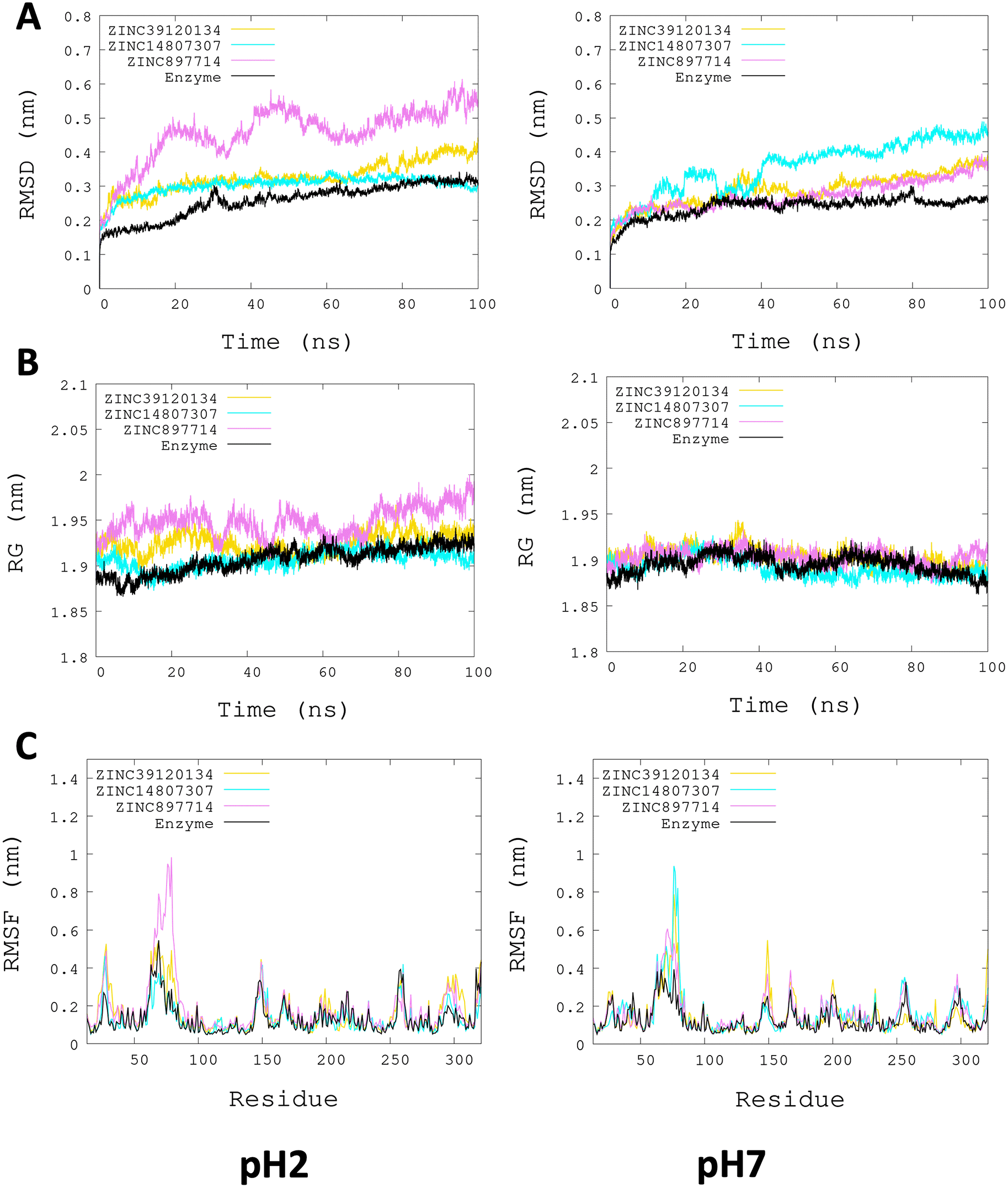
More significant conformational changes of ARG enzyme are shown at pH 2.0. (A) RMSD plot of ChainA concerning the whole protein. (B) RG analysis. (C) RMSF per residue of backbone.
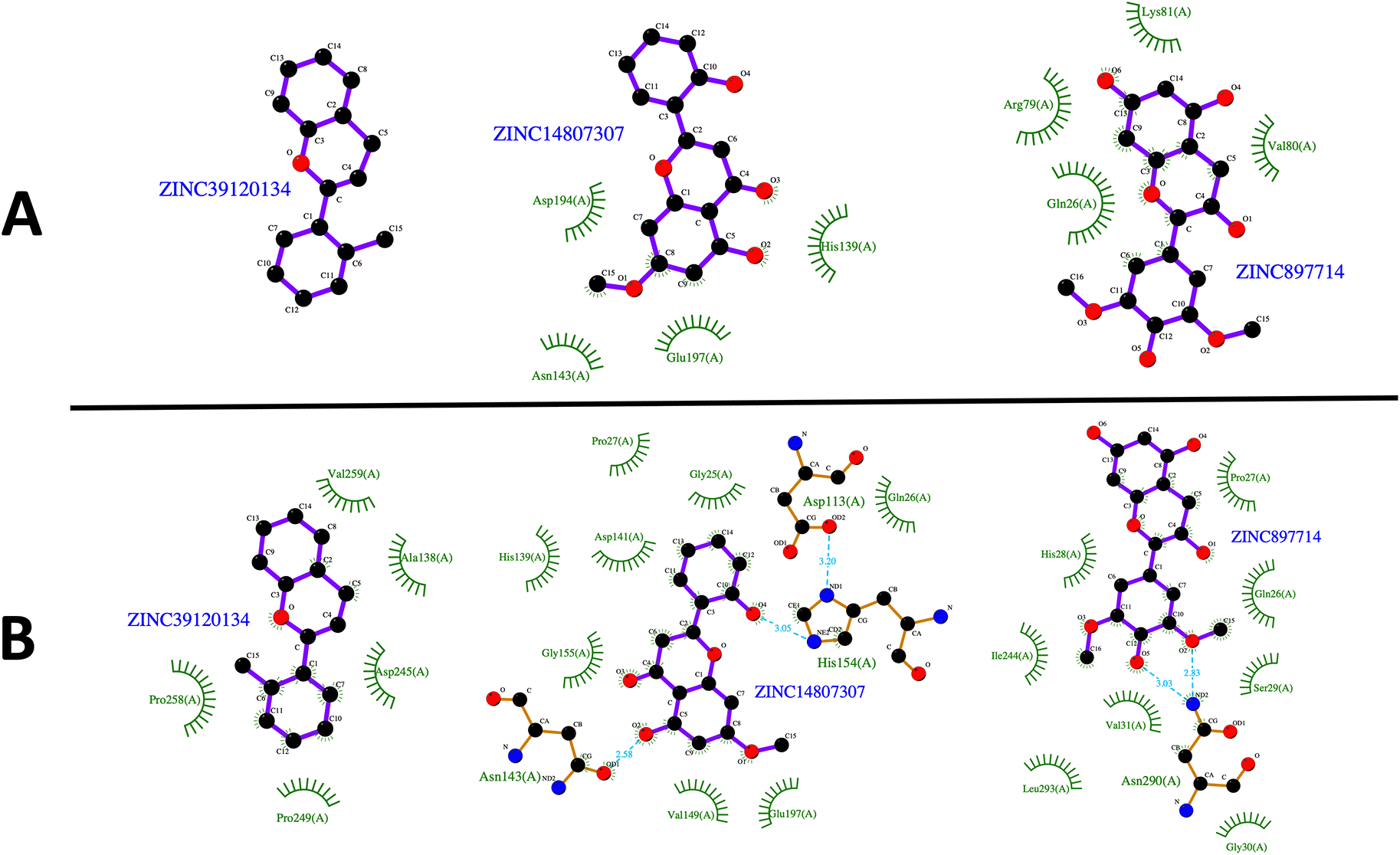
(A) Last frame at pH 2.0. and (B) Last frame at pH 7.0. Green represents the hydrophobic interaction between enzyme-ligand, color sky blue represents the hydrogen bond interaction.
The binding free energy analysis of pH 2.0 and pH 7.0 from the frames of each simulation is shown in Table 2. The propitious energetic contribution with a binding free energy of -28.59 kcal/mol (ZINC897714/pH2) maximum and -14.07 kcal/mol (ZINC14807307/pH2) minimum were obtained. The estimated phase-gas binding free energy (ΔGgas) provided the highest energy contributions for ZINC897714 in both pHs. Contrary, the van der Waals energies (ΔEvdW) provided the highest energy contributions at pH 2.0 in ZINC897714.
It is well understood that hydrophobic interactions favorably contribute to binding. The electrostatic energies (ΔEele) contributed positively to the binding enzyme-ligand, which the best energy was -24.76 kcal/mol (ZINC897714/pH2). Despite this, the solvation energies (ΔGsolv) offset the negative electrostatic interactions, thus unfavorably contributing to the binding of ZINC897714 to ARG in both pHs (ZINC897714/pH2 = 29.64 kcal/mol and ZINC897714/pH7 = 22.18 kcal/mol). These results show that the protonation states at a given pH can positively or negatively favor the enzyme-ligand binding, where it is expected that at a pH above 7.0 the enzyme-ligand binding can be increased.
In an attempt to improve the enzyme-ligand binding energy analysis, the FEP approach was used, which estimates the difference in free energy between two states (A state and B state) by slowly change from one state to another. A state corresponds to the initial state of free energy and B state corresponds to the final state. This study sampled 20 microstates with a time of 20 ns for each microstate; the results are presented in Table 3. Herein, it is observed that, at both pHs, the best compounds occurred in the following order: ZINC14807307 > ZINC897714 > ZINC39120134. On the other hand, the compounds ZINC14807307 and ZINC897714 are shown to be stable at pH 2.0 conditions.
The World Health Organization (WHO) considers leishmaniasis to be one of the major neglected global diseases and responsible for millions of disability-adjusted life years (DALYs), representing one of the top burdens among the neglected tropical diseases.63 Worldwide, 13 countries have a high burden of VL (Bangladesh, China, Ethiopia, Georgia, India, Kenya, Nepal, Paraguay, Somalia, South Sudan, Spain, Sudan, and Uganda), and 11 have a high burden of TL (Afghanistan, Algeria, Colombia, Iran, Morocco, Pakistan, Peru, Saudi Arabia, Syrian Arab Republic, Tunisia, and Turkey), while Brazil has a high burden of both clinical forms.64 Thus, TL treatment choice is based on the clinical presentation and infecting species, while any person with VL signs and symptoms and a verified diagnosis warrants chemotherapy.65 The range of currently available drugs for treating leishmaniasis is relatively small and it includes repurposed molecules, such as amphotericin B, miltefosine, and paromomycin; while few new drug candidates reached clinical trials in the last decades.66,67 For these reasons, the investigation of new therapies has been very active recently, and a wide range of compounds have been identified as potential hits and leads.68 The unique and vast chemical diversity of NPs places them as a major component of the biologically relevant chemical space,69 while NP classes like alkaloids, coumarins, flavonoids, lignans, neolignans, quinones, and terpenoids have demonstrated anti-leishmanial activity.70 Several of these that target Leishmania ARG have been investigated for their potential as new drug candidates, although quercetin,71–73 catechin, (-)-epicatechin, (+)-syringaresinol, isoquercetin, quercitrin, resveratrol, and cinnamic acid derivatives had shown in vitro efficacy.31,33,74 Additionally, certain NPs had demonstrated favorable in vivo effectivity, including epigallocatechin gallate,75 gallic acid,76 rosmarinic acid,77 and quercetin.78,79 The equilibrium between biological activity and pharmacological qualities is one of several aspects, nevertheless, that restricts the translation of NPs into commercial drugs.80,81 In silico based drug repositioning potential for discovering new applications for existing drugs and for developing new drugs in pharmaceutical research and the industry has gained importance82,83; whereas, in the chemical structure and molecule information approach, the structural similarity is incorporated with molecular activity and other biological information to identify new associations.84
The present work aimed to apply CADD approaches to select analogs to NPs with known anti-leishmanial and anti-ARG activities; although results of the quercetin analogs, the anthocyanin malvidin (ZINC897714; PubChem CID: 159287), and the flavone echioidinin (ZINC14807307; PubChem CID: 15559079) showed favorable binding affinity to L. infantum, L. mexicana, and L. braziliensis ARG and no predicted toxicity. Besides that, in the ARG super-family, the active site is conserved in all organisms, which includes the coordination of divalent metal Mn2+,85 and differences between the parasite and its human homolog have been described,86,87 highlighting the possibility to target selectively the parasite enzyme. However, recently, cinnamides88 and 1-phenyl-1H-pyrazolo[3,4-d] pyrimidine synthetic derivatives89 have been described as potential selective inhibitors of parasite ARG and have shown in vitro anti-leishmanial activity. A major bottleneck of drug discovery for leishmaniasis was aimed at the in silico workflow proposed, which is that compounds must show activity in the acidic environment of the phagolysosome90; thus, the analyzed compounds in this work showed stable enzyme-ligand interaction and favorable binding free energy at pH 2.0 in MDS analysis. However, when taking into consideration the target product profile (TPP), proposed by the Drugs for Neglected Diseases initiative (DNDi), which includes regard for the oral route of administration for new candidates,91 both ADME profiles showed the potential for oral route administration and high bioavailability, but only malvidin results have been ratified by experimental studies published elsewhere.92–94 Furthermore, malvidin has shown the potential to be an antioxidant, anti-hypertensive, anti-inflammatory, anti-obesity, anti-osteoarthritis, anti-proliferative, and anticancer drug candidate,95–99 whereas to the best of our knowledge no research has been published studying the potential pharmacological activity of echioidinin. Anthocyanins are commonly found in many plants, while the most common types are cyanidin, delphinidin, pelargonidin, peonidin, petunidin, and malvidin, which are distributed in fruits and vegetables in 50%, 12%, 12%, 12%, 7%, and 7% proportions, respectively.100 These molecules are more stable at a lower pH solution, and in such conditions the flavylium cation formed enables the anthocyanin to be highly soluble in water.101 The physicochemical properties offered by anthocyanins should be considered of interest for anti-leishmanial drug discovery since the parasite is adapted to live in parasitophorous vacuoles of infected macrophages in mammalian hosts, where it survives, proliferates, and is responsible for the development of the active disease.102 Recently, the anthocyanidin profile of Arrabidaea chica has been examined and its anti-leishmanial activity analyzed,103 and carajurin (PubChem CID: 44257040) showed the highest activity against the intracellular parasites, altering all parameters of in vitro infection.104 Additionally, it has been shown that carajurin leads to a decrease in the mitochondrial membrane potential, an increase in ROS production, and cell death by late apoptosis in L. amazonensis.105 Furthermore, flavones showing anti-leishmanial potential have been described in the literature,106 whereas apigenin (PubChem CID: 5280443) and luteolin (PubChem CID: 5280445) have shown the potential of inhibiting the growth of L. amazonensis.107
Limitations of the present study should be also mentioned, such as the protein dynamics and complex stabilities with MDS lasting within nanoseconds scales (0-100 ns), while most structural dynamics and biological activities of proteins occur within timescales of microseconds and milliseconds.108 Even so, complex dynamics and interactions between enzymes and ligands have been reported using nanosecond timescales.109,110 Additionally, the work did not include in vitro or in vivo validation. It is important to note that anti-leishmanial in vitro assays have drawbacks, including metabolic differences between the amastigote and promastigote stages,111 variations in drug effectiveness and susceptibility among parasites isolated from patients,112 and a variety of biochemical pathways linked to drug-resistant phenotypes in the parasite,113,114 which can lead to false positive results. Additionally, numerous animal models are used in the validation tests for VL and TL drug candidates; however, due to insufficient translation to human disease, their predictive value is frequently low. Furthermore, reliable main models for VL are frequently employed, including Syrian golden hamsters and BALB/c mice,115,116 while there are no validated animal models for TL since different species experience varied clinical symptoms, and current models lack human characteristics such as pathophysiology, symptomatology, and treatment response.117
In the first screening, this work identified three substances with natural products structural analogs with potential effects against Leishmania ARG using in silico analysis from the available data and research of natural products found in databases. The substances were: ZINC39120134 (3,4-dihydro-2-(2-methylphenyl)-(9CI)), ZINC14807307 (echioidinin) and ZINC897714 (malvidin), where the most suitable compounds were ZINC14807307 and ZINC897714, showing favorable binding affinity to L. infantum, L. mexicana, and L. braziliensis ARG, no potential toxicity and stability at pH 2.0; important factors due to the acidic environment of the phagolysosomes of mammalian hosts. Taking into consideration that the oral bioavailability of malvidin has experimental data published and that its pharmacological potential has been widely studied, the results presented in this work warrant further in vitro and in vivo studies using malvidin to confirm its potential as a drug candidate against leishmaniasis.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)