Keywords
muscular dystrophy, Duchenne, Facioscapulohumeral, Bethlem, Limb-girdle, strategies, Merosin-deficient, Emery Dreifuss.
muscular dystrophy, Duchenne, Facioscapulohumeral, Bethlem, Limb-girdle, strategies, Merosin-deficient, Emery Dreifuss.
1. We reviewed the discussion again so that the types of dystrophies were clear.
2. We mentioned in the comment section the fact that we found a small number of cases for muscular dystrophy, as we followed to a hospital-based reference.
3. We clarified in the discussion section that the prevalence reported in this study are related to a population of patients who were tested for multiple indications including CPK levels. Due to limitations of the study, we cannot talk of a population prevalence but rather of a hospital prevalence.
4. We explained in the Study strengths and limitations section that patients in our country do not generally have access to molecular tests, so we perform a clinical characterization based on CPK levels, electromyography, and physical examination.
5. We established in the discussion section that we do not have anything novel to the existing knowledge about muscular dystrophies, but it is considered for general physicians to have a broad perspective and the different causes of an elevated CPK when they treat them in the emergency room, as it will allow patients with dystrophy to be diagnosed earlier. In addition, CPK was used in this study mostly to search for patients with probable dystrophy or confirmed muscular dystrophy.
6. We summarized two paragraphs concerning inflammatory myopathies from the discussion section.
7. We moved a paragraph from the Results section to the Methods section to better explain how we performed the patient selection process.
8. We added missing prevalence from the study in a new paragraph in the Results section.
9. We added a new paragraph in the Conclusion section.
See the authors' detailed response to the review by Prajnya Ranganath
Creatine phosphokinase (CPK) is an enzyme that catalyzes phosphocreatine to facilitate the release of the energy required by the muscle for its contraction.1 This enzyme is widely distributed in all tissues with high-energy demand. Approximately, 70% of CPK is found in the musculoskeletal system, 20-30% in the cardiac muscle, and 5-10% in the nervous system.2 CPK presents in three different molecular forms depending on the tissue: BB, MB, and MM.2 Blood levels of CPK are mainly from the muscle MM isoenzyme. The blood concentration of CPK depends on multiple factors such as gender, race, age, muscle mass, and physical activity.3,4 Normal values are: white female <= 325 IU/L, white male <= 504 IU/L, black female <= 621 IU/L, and black male <= 1200 IU/L.5 In general, the term CPKAEMIA is considered when CPK levels are greater than 1.000 IU/L or 1.5 times the upper limit of normal.5
For transient CPK elevations, multiple causes must always be ruled out in the initial diagnostic confrontation. Intense physical exercise, trauma, generalized tonic-clonic seizures, acute psychosis, systemic connective tissue diseases, renal or cardiac failure, viral diseases that cause myopathies such as influenza, coxsackie, adenovirus, prostate cancer, celiac disease, obstructive sleep apnea, moderate to severe hypothyroidism, drugs such as statins, fibrates, antiretrovirals, beta-blockers, clozapine, angiotensin II receptor antagonists, hydroxychloroquine, isotretinoin, and colchicine; neuromuscular diseases such as Guillain Barré, amyotrophic lateral sclerosis, myopathy due to the use of corticosteroids, hyperthyroidism, collagen diseases, alcoholism, procedures such as intramuscular injections and electromyography have been described as possible etiologies.6–12 Besides that, the most frequent causes of CPK elevation are genetic diseases such as muscular dystrophies, congenital myopathies, channelopathies, mitochondrial myopathies, and myotonic dystrophies, although exercise can also temporarily increase CPK levels due to rhabdomyolysis.2,13
Muscular dystrophies are classified according to their genetic transmission mechanism, either autosomal dominant or recessive, and those linked to the X chromosome. It can also be done based on the structural and functional deficiency of the protein complex as dystrophinopathies (Duchenne muscular dystrophy and Becker), laminopathy (LGMD 1B and Emery dreifuss), sarcoglycanopathies (LGMD 2C, 2D, 2E, 2F), dysferlinopathies (LGMD 2B), calpainopathies (LGMD 2A), myotilinopathies (LGMD 1A), and caveolinopathies (LGMD 1C).14,15
Dystrophies can affect different stages of life. Its onset depends on the type of dystrophy: congenital or adulthood, present between 20 and 30 years of age, and it can even have such mild clinical manifestations that consultations for the disease are made only until an older age. In some cases a phenomenon called “anticipation” may occur, that is, when the clinical manifestations may occur earlier in the children than in their parents; or spontaneous mutations, such as the Duchenne type, which occur in children without a family history of the disease.
Regarding affected muscles, each muscular dystrophy compromises similar or different muscular types. Duchenne and Becker muscular dystrophy affect striated and cardiac muscles with associated increased CPK levels between 10 and 50 times the upper limit of normal; facioscapulohumeral muscular dystrophy affects striated muscle and respiratory muscles with associated increased CPK levels between 1 and 10 times the upper limit of normal; Bethlem muscular dystrophy affects only striated muscle with associated increased CPK levels between 1 and 10 times the upper limit of normal; limb-girdle muscular dystrophy affects striated, cardiac and respiratory muscles with associated increased CPK levels between 1 and 50 times the upper limit of normal; Emery Dreifuss muscular dystrophy affects striated and cardiac muscles with associated elevated CPK levels from 1 to 10 times the upper limit of normal.
Early clinical complications of CPK elevation include liver dysfunction and arrhythmias; late complications include disseminated intravascular coagulation (DIC) and acute kidney injury (AKI).13 Thanks to new advances in gene therapy, there are current trials targeting to restore damaged genes in different dystrophies (Duchenne, limb-girdle, and Emery Dreifuss). These new therapies use adeno-associated virus to infect damaged tissues and deliver a cDNA copy of the functional gene. The results have not been published out yet but there are early promising reports, with studies in mice having demonstrated therapeutic efficacy.15–18
Less common muscular dystrophies are congenital myopathies such as myosin storage myopathy 7A, which is an autosomal dominant disease with a heterozygous mutation in the MYH7 gene.19 It has phenotypic variability with an age onset ranging from early childhood to late adulthood. Affected individuals have proximal muscle weakness of the upper and lower limbs, and distal muscle weakness of the lower limbs, resulting in gait difficulties and scapular winging (scapuloperoneal myopathy).19 The severity is also variable, and CPK levels may be normal or elevated.19 The disease is usually slowly progressive and most patients remain ambulatory. Skeletal muscle biopsy can show a nonspecific myopathic pattern.19
Lastly, acute viral myositis is another cause of elevation in CPK levels that could mask muscular dystrophies. According to Cardin et al, it constitutes a syndrome that generates musculoskeletal impairment after upper airway disorders that results in temporarily limited ambulation, muscle pain, and lower-limb weakness, especially in the calves and thighs.20 The disease management is usually symptomatic of respiratory and musculoskeletal symptoms including analgesics or anti-inflammatory drugs.20
Regarding the comments above, we aimed to establish clinical and molecular characteristics of patients with increased CPK levels and muscular dystrophies in our region to facilitate diagnosis and follow-up on patients with suspected muscular dystrophies.
A cross-sectional study was made using a retrospective search of patients attended in Comfamiliar Risaralda between 2010 and 2021. The study included patients from both genders and all ages who presented with CPK levels greater than 500U/L, and diagnosis of polymyositis, myoclonus, myopathy, and muscular dystrophy.
A database analysis was carried out from 2010 to 2022 of 5219 patients treated in a fourth-level care institution in the Eje Cafetero region. As shown in Figure 1, 1949 patients were excluded with other causes of elevated CPK levels such as hypothyroidism, acute myocardial infarction, cardiac insufficiency, rhabdomyolysis, seizures, drugs including statins, fibrates, and nonclear causes.
After reviewing medical records, 98 patients did not meet the inclusion criteria for the investigation due to other causes of CPK elevation such as drowning (submersion injuries), perinatal asphyxia, cranioencephalic trauma caused by a car accident, trauma caused by white weapons, and electrostatic discharge (Figure 1).
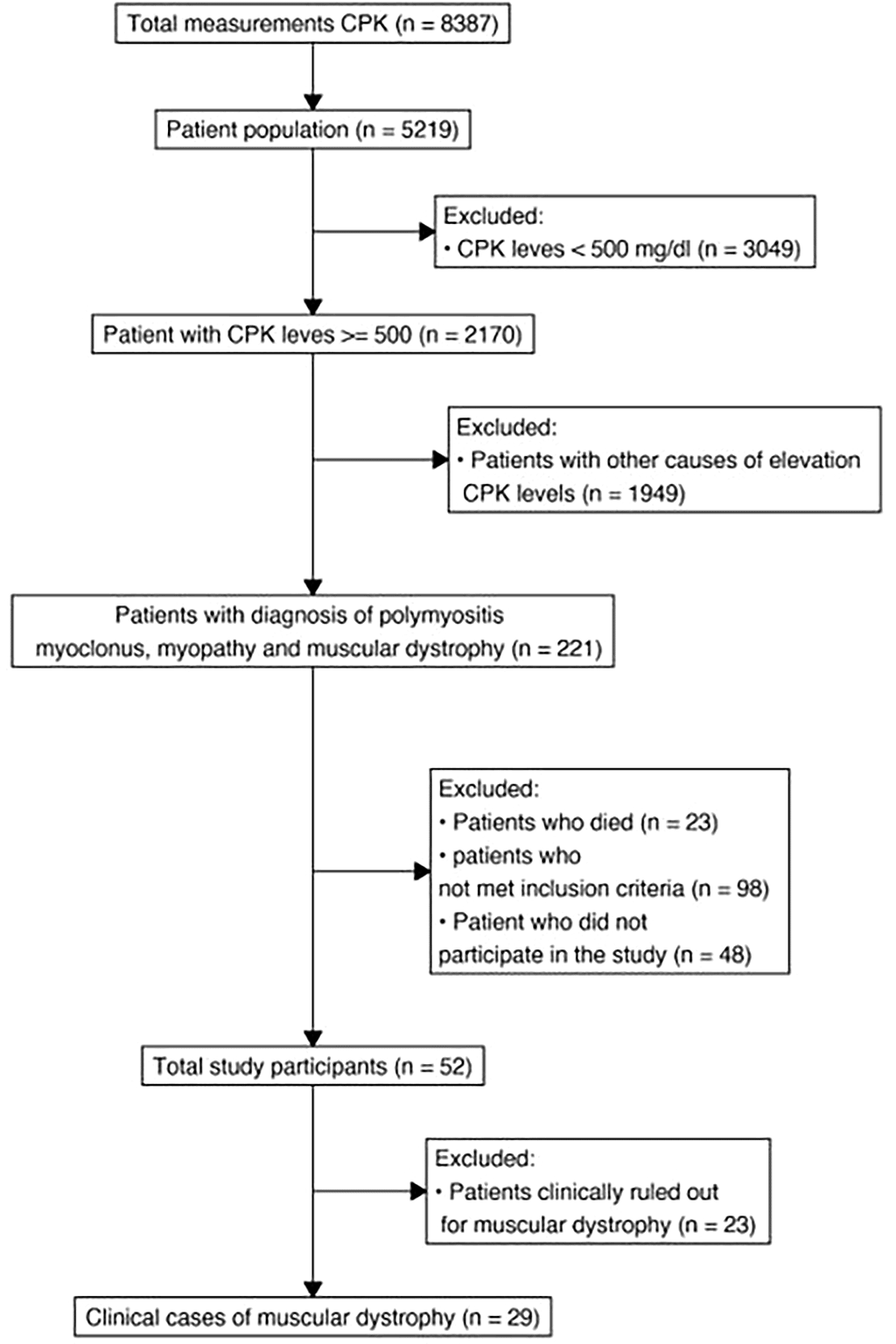
The remaining 52 patients underwent medical consultation and 23 were excluded because some did not meet the clinical criteria for any muscular dystrophy but for other causes of transient elevation of CPK levels, and others manifested discontinuation in the study (Table 1). Finally, the patients underwent a second medical consultation by a geneticist to corroborate and classify the remaining 29 patients.
In an initial instance of the study, a retrospective institutional active search was made through text mining in laboratory databases for all CPK measures carried out by the Comfamiliar Risaralda laboratory between 2010 and 2021. After that, the results were purified and organized under the criteria of registered CPK elevation, and again the search language in databases was made to extract new confirmed or repeated diagnoses that could explain the CPKemia.
Data collection was implemented through a study-specific case report form (CRF). A detailed data validation plan that identified missing data, out-of-range data and other data inconsistencies in the form of revision checking was implemented on the platform before the start of the study.
The collected information was entered into the RedCAP platform, where the CRF was previously located with validation and automated fields if applicable.
Discrepancies in the data were reported to the team as queries. Access to the database was restricted through password protection to authorized data management personnel.
Descriptive statistics were obtained for numerical variables, including measures of central tendency such as mean and median. Descriptive statistics for categorical variables included a tabulation of frequencies with counts and percentages.
The estimation of prevalences was carried out employing Bayesian inference since they allow a greater gain in precision in the estimation of parameters with values below 10%, as well as it was carried out utilizing a beta-binomial model under the binom package of R software. Statistical analyzes were also performed in R software.
We found 29 patients with clinical muscular dystrophy in our hospital-based registry, with a mean CPK value of 3231.241, a standard deviation of 5019.543, and a maximum of 22670. We found a combined prevalence of all muscular dystrophies of 4.2 per 100.000 patients attended in our hospital base, Duchenne muscular dystrophy of 0.6 per 100.000, limb-girdle muscular dystrophy of 0.6 per 100.000, facioscapulohumeral dystrophy of 0.5 per 100.000, Bethem dystrophy, type 2 Emery Dreifuss muscular dystrophy, merosin-deficient muscular dystrophy and myosin storage disease of 0.1 per 100.000.
Several studies have described the different types of muscular dystrophy. However, there is poor evidence analyzing multiple muscular dystrophies within the same population. This leads to heterogeneity in studies regarding prevalences, clinical manifestations, and a good genotype-phenotype correlation. In our study, we found patients with Duchenne and Becker muscular dystrophy, facioscapulohumeral muscular dystrophy, limb-girdle muscular dystrophy, Emery Dreifus muscular dystrophy, merosin-deficient muscular dystrophy, and autosomal dominant congenital myopathy 7A (Table 2). Therefore, to identify a patient with a particular type of disease, clinicians should follow a diagnostic sequence not only for differentiating muscular dystrophy from other inflammatory myopathies but also to discriminate the specific type.
| N = 29 (%) | Prevalence (× 1000)** | CrI95%* | |
|---|---|---|---|
| Bethem dystrophy | 1 (3.4%) | 0.28 | 0.02-0.89 |
| Facioscapulohumeral dystrophy | 5 (17.2%) | 1.0 | 0.36-2.0 |
| Unclassified dystrophy | 2 (6.8%) | NA | NA |
| Becker muscular dystrophy | 3 (10.3%) | 0.67 | 0.16-1.5 |
| Limb-girdle muscular dystrophy | 5 (17.2%) | 1.0 | 0.36-2.0 |
| Duchenne muscular dystrophy | 7 (24.1%) | 1.4 | 0.6-2.6 |
| Type 2 Emery Dreifuss muscular dystrophy | 1 (3.4%) | 0.28 | 0.02-0.89 |
| Merosin-deficient muscular dystrophy | 1 (3.4%) | 0.28 | 0.02-0.89 |
| Myotonic dystrophy | 2 (6.8%) | 0.47 | 0.079-1.2 |
| Autosomal dominant congenital myopathy 7A | 1 (3.4%) | 0.28 | 0.02-0.89 |
In our study, we analyzed the clinical and molecular characteristics of 29 patients with muscular dystrophy in Colombia. According to Mercuri and Muntoni, the prevalence of Duchenne muscular dystrophy is 8-29 per 100 000 boys; Becker muscular dystrophy has a prevalence of 7-29 per 100 000 boys; limb-girdle muscular dystrophy has a prevalence of 0.8-5.7 per 100 000 inhabitants; myotonic dystrophy has an estimated prevalence of 10-6 per 100 000 men, followed by facioscapulohumeral muscular dystrophy with a prevalence of three per 100 000 men, and myosin storage disease with noncurrent reported prevalence.14 We found a combined prevalence of all muscular dystrophies of 4.2 per 100.000 among patients treated in our hospital base, Duchenne muscular dystrophy of 0.6 per 100.000, limb-girdle muscular dystrophy of 0.6 per 100.000, facioscapulohumeral dystrophy of 0.5 per 100.000, Bethem dystrophy, type 2 Emery Dreifuss muscular dystrophy, merosin-deficient muscular dystrophy and myosin storage disease of 0.1 per 100.000. These prevalences are lower than those reported in the literature and can be explained due to a bias regarding the filter we used to find the patients. We included patients from both genders and all ages who presented with a diagnosis of polymyositis, myoclonus, myopathy, and muscular dystrophy. However, some other suspected cases could have been recorded with different diagnoses, due to misdiagnosis or underdiagnosis, leading to underestimation.
Duchenne muscular dystrophy age onset of symptoms is in early childhood around the age of 5 years, it is rapidly progressive and most patients lose ambulation around 10-15 years old, leading them wheelchair dependent.21 Becker muscular dystrophy symptoms can start around 10-20 years old but in some cases, permanent asymptomatic until adulthood even >30 years, and remain ambulant even until their 60s.21,22 In our study, we found that most patients had similar features as previously reported in the literature. Three patients were revealed with the c.6439-?_6912+?del p.(Glu2146_Val2304del) mutation who presented with a mild phenotype of Becker muscular dystrophy. At the moment of consultation, they had a mean age of 48 years (SD 40-54). All of them presented with hyporeflexia, mild muscle weakness, age onset of symptoms between 10 and 20 years old, and any of the three patients were wheelchair dependant. Greer et al. reported this same mutation in a patient with Becker muscular dystrophy who lost ambulation by the age of 15 years.23 They suggest that patients with apparently identical exonic deletions are almost certainly going to have different genomic breakpoints and therefore will be missing different intronic regions, and potentially, splicing motifs resulting in different phenotypes.23 Vengalil et al. also identified that patients with deletions of exons 45-47 are clinically related to the development of cardiomyopathy and earlier wheelchair dependence.24 Further studies analyzing genotype-phenotype correlation in patients with muscular dystrophy would be helpful to better understand the pathogenesis, and predict phenotypes.
Facioscapulohumeral dystrophy has an onset around 10-20 years old. Still, there are some unusual cases where the symptoms are present at the moment of birth, and some others remain asymptomatic their whole life. By the age of 20 years, 50% of the cases have developed symptoms, and eventually, 20% of the affected require a wheelchair, which is consistent with our study where patients with facioscapulohumeral dystrophy developed symptoms after 20 years old.25
In Bethlem muscular dystrophy, the symptoms can range from congenital to mid-adulthood, been the congenital cases extremely rare.26 The progression is slow and more than 2/3 of the affected older than 50 years require supportive means (cane, crutches, wheelchair) for outdoor mobility.27 This is consistent with our study where we found one patient with Bethlem muscular dystrophy who presented as a congenital case that also had supportive means for outdoor mobility.
Limb-girdle muscular dystrophy is an extremely heterogeneous group. Over 30 distinct subtypes have been identified, in which the onset of symptoms is at any age with some severe congenital cases and some mild cases starting in adulthood; the severity is also variable.28 As stated in the literature, we found four patients with variable phenotypes ranging from mild disease with onset of symptoms over 20 years old to more severe cases with early onset of symptoms in childhood.
Emery Dreifuss muscular dystrophy onset of symptoms begins in the first two or three decades of life.29 The progression is slow, and loss of ambulation can occur in the autosomal dominant variant but is rare in the X-linked variant.30 We found one patient with Emery Dreifuss muscular dystrophy whose symptoms corresponded with those reported in the literature.
Merosin-deficient muscular dystrophy is a severe type with symptoms present at the moment of birth, with neonatal profound hypotonia, poor spontaneous movements, and respiratory failure;31 most affected children do not acquire independent walking. It has been reported that only 15% of individuals acquired independent ambulation,32 and only a few patients gained the ability to walk with assistance but subsequently lost the ability.33 In our study, we identified one patient with Merosin-deficient muscular dystrophy who presented with profound hypotonia since birth and was diagnosed at two months old with CPK levels and molecular test. Currently, this patient can adequately sit but has problems with gait. These features correspond with phenotypes previously reported in the literature but without respiratory compromise.
We also found a patient with autosomal dominant congenital myopathy 7A with a phenotype that presented in early childhood at 14 months old, with significant proximal and distal muscle weakness. In addition, the patient presented with a severe restrictive pulmonary pattern, and a myopathic pattern in electromyography without cardiac alterations. Currently, at his adult age, this patient has proximal and distal quadriparesis and hypoesthesia of the left hemibody. These features correspond with the previously reported in the literature.
On the other hand, 12 patients with muscular dystrophy remained unclassified despite their clinical suspicion and after undergoing molecular tests. As previously exposed, patients with similar exonic deletions are going to have different genomic breakpoints. The number of genetic alterations that can be involved in muscular dystrophies is huge. Therefore, performing a single molecular test sometimes is not enough to identify the specific mutation related to the phenotypic presentation of the patient. Table 3 shows a brief review of the clinical and paraclinical approach to the different types of muscular dystrophy. Table 4 shows the detailed classification of each type of muscular dystrophy.
Barohn et al. propose a series of steps regarding the patient’s symptoms by establishing which muscle-related symptoms patients demonstrate; determining the temporal evolution of the symptoms; interrogating if there is a family history of a myopathic disorder; finding out if there are precipitating factors that trigger episodic weakness or stiffness; asking if there are associated systemic symptoms or signs; and lastly, identifying the distribution of Weakness.34 After completing the 6 steps, clinicians could now attempt to classify a myopathic disorder according to the patterns of muscle weakness. The findings on the physical examination, particularly the distribution of muscle weakness, should provide additional information in determining the correct diagnosis.34
Subsequently, CPK levels are extremely helpful for the evaluation of patients with a suspected myopathy. The CPK levels are usually elevated in most patients with muscle disease but may be normal in slowly progressive myopathies.34 Depending on the degree of CPK levels, it can be useful in distinguishing different forms of muscular dystrophy. As in the case of Duchenne muscular dystrophy, CPK levels can be elevated from 10 to more than 100 times the upper limit of normal, whereas in Facioscapulohumeral muscular dystrophy, the CPK levels are expected to be elevated from 1 to 10 times the upper limit of normal. We also must rule out nonmyopathic factors that can alter CPK levels such as profound muscle wasting, corticosteroid administration, collagen diseases, alcoholism, or hyperthyroidism.34
Later, we should look for electromyography and nerve conduction studies. These studies can help to distinguish primary neuropathic from myopathic disorders by confirming that the muscle is the correct site of the lesion and that weakness is not the result of an underlying motor neuron disease, neuropathy, or neuromuscular junction disorder.34 However, their sensitivity and specificity are low. Normal results provide evidence of a nonsevere neuromuscular disorder.5
A few years ago, if the electrodiagnostic features suggested a myopathic pattern, a muscle biopsy was recommended. Additionally, we can now diagnose muscular dystrophies through molecular tests without performing muscle biopsies in all suspected patients. Table 5 shows the patterns of muscular dystrophy applied to the 29 participants of the study.
An adequate diagnosis is done on a clinical basis and confirmed by laboratory tests, muscle enzyme concentrations, autoantibodies, electromyography, and muscle biopsy.35 Depending on the type of muscular dystrophy, some complications such as restrictive lung disease, cardiomyopathy, scoliosis, corticosteroid side effects, and psychosocial issues can generally lead professionals to focus only on the direct acute cause of the signs and symptoms and not to search for a long-term systemic etiology like muscular dystrophy.36 Therefore, clinicians need to figure out that patients presenting with some acute diseases, especially in recurrent cases, might also be associated with muscular dystrophy to keep in mind.
The strengths of this study are the diagnostic sequence to classify muscular dystrophy, and the fact that this was the first study exploring multiple types of muscular dystrophy within the same population in this region. Regarding limitations, a bias in the filter we used to find the patients could have allowed for underestimation in some frequencies. Some prevalences that we found need confirmation in further studies. On the other hand, in our country patients do not generally have access to molecular tests, so we perform a clinical characterization based on CPK levels, electromyography, and physical examination. We hope in further studies to have the possibility of performing molecular confirmation as well.
Although muscular dystrophies consist of a heterogeneous group of neuromuscular diseases, there are still clinical and paraclinical features that can help physicians to detect any particular case and perform a good approach and follow-up. In our region, many patients with muscular dystrophy remain underdiagnosed or misdiagnosed, which in consequence it compromises their prognosis without adequate treatment. Therefore, our diagnostic sequence will facilitate physicians to identify any of the most frequent muscular dystrophies.
Moreover, many patients with muscular dystrophies can remain without optimal treatment due to inaccurate diagnosis or confusion with other inflammatory myopathies such as polymyositis, dermatomyositis, and inclusion body myositis.21 In this study, 13.1% of patients diagnosed these other myopathies ended with confirmed muscular dystrophy after performing a detailed revision of their clinical histories and re-consultation of such patients. For this reason, we suggest implementing at least a 6-month follow-up to rule out newly o persistently elevated CPK levels that might indicate a muscular dystrophy.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
{kind=link}
Comments on this article Comments (0)