Keywords
network biology, module detection, graph clustering, graph representation learning, node embedding, graph neural network, machine learning
This article is included in the Bioinformatics gateway.
This article is included in the Machine Learning in Genomics collection.
network biology, module detection, graph clustering, graph representation learning, node embedding, graph neural network, machine learning
Graphs provide ubiquitous powerful representations of interacting components in many complex systems.1 In molecular biology, graphs are used to represent interactions between molecular components; for example, protein-protein interactions represent interactions between proteins, while gene regulatory networks describe interactions between transcription factor proteins and genes. A common property of graphs of complex systems, including molecular networks, is the presence of community structures or modules, defined by groups of densely interacting nodes. In protein-protein interaction networks, such modules often correspond to key biological processes needed for cells to accomplish different tasks.2 Disruptions to module connectivity have been implicated in many diseases.1 Therefore, the accurate detection of these modules is an important problem in the analysis of molecular networks.3 Graph representation learning (GRL),4 which aims to embed graph nodes in a -dimensional space (Figure 1), offers a powerful framework for performing machine learning tasks on graphs, including clustering.5 Both deep and shallow graph representation algorithms exist; however, deep representation learning methods have been designed and tested on non-biological networks such as social networks, citation networks and recommendation systems6–9 or only tested on link prediction and node classification tasks.10 It is unclear if deep graph representation learning methods are beneficial for module detection of molecular networks.
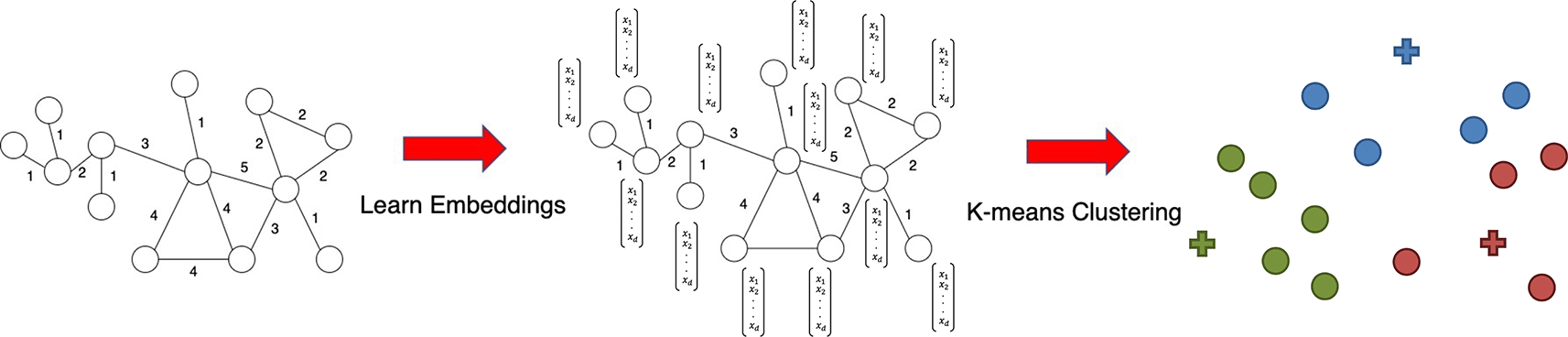
A GRL algorithm converts graph structure into node embeddings denoted as vectors for each node. A standard clustering method, such as K-means clustering, can be applied to embeddings to obtain group clusters.
To address this need, we benchmarked several state-of-the-art shallow and deep learning GRL frameworks for the task of module detection on both real molecular networks and synthetic benchmark networks. Our benchmark included three shallow GRL methods: Graph Laplacian embedding,11 DeepWalk,12 and node2vec13 and two deep GRL algorithms: Variational Graph Auto-Encoder (VGAE)7 and GraphSAGE.14 We evaluated the methods on one simulated and three real world molecular network datasets. Two of the three real network datasets captured the impact of single nucleotide polymorphisms (SNPs) identified from genome wide association studies (GWAS). We used multiple clustering metrics to evaluate the quality of the inferred clusters including modularity index, silhouette index, adjusted mutual information index (when cluster labels were available) and enrichment of gene ontology processes for modules inferred on the molecular networks. Our results show that on simulated benchmarks, deep GRL methods offered some advantages over shallow methods, however, on real networks we did not see a clear advantage of deep GRL methods over shallow GRL methods for detecting modules in biological networks.
We applied five different representation learning algorithms to simulated and cell type-specific gene interactions that we previously generated.15
Lancichinetti-Fortunato-Radicchi (LFR) benchmark graphs
We evaluated all the algorithms on a synthetic network based on the LFR graph generator. We chose to use LFR benchmark graphs16 as they can support larger graphs and capture the distribution of node degree and community sizes of real-world networks, which often follow the power law distribution. We used the graph generator to obtain weighted undirected graphs with ground-truth non-overlapping communities.
The LFR benchmark has a number of parameters: (,,,,,,,,). is the number of nodes in the whole graph, is the average degree of nodes, is the exponent value for the power-law distribution of node degrees, is the exponent value for the power-law distribution of community sizes, and is the mixing the parameter controlling the percentage of the degree of each node interconnecting from its own community to another community. When the mixing parameter is low, there is a higher chance for an edge to stay within one community; while when it is high, there will be more interconnected edges between two communities than edges with two ends in the same community. The last four parameters , , , are the lower and upper bounds of node degree and community size when constructing synthetic networks.
By setting different parameters in the LFR package, we can generate networks of different topological properties and compare various node embedding techniques. Based on the statistics of the 55 cell-type specific networks, we set the number of nodes , the average degree of nodes , the exponent for degree distribution , the exponent for community size distribution , and the mixing parameter . We left the bounds of node degree and community size as default. For each parameter setting, we generated five sets of weighted synthetic networks. Since some of our GRL algorithms take unweighted graphs, we derived unweighted graphs by keeping edges with weights higher than the average weight. In total, there are eight groups of synthetic network datasets for each parameter setting with five networks each in total (Tables 1 and 2).
The unweighted networks comprise edges with weights larger or equal to the average edge weights in weighted ones.
These values can be used to compare with the optimal k value detected when applying K-Means in each algorithm.
| Average # of node communities | Average # of node communities | ||
|---|---|---|---|
| k = 10 | 88.8 | k = 10 | 96.6 |
| k = 25 | 34.6 | k = 25 | 33.6 |
| k = 50 | 18 | k = 50 | 18.6 |
| k = 100 | 9.2 | k = 100 | 9.4 |
Cell-type specific networks
The cell-type specific networks consist of 55 gene networks capturing protein-protein and transcription factor (TF) gene regulatory interactions in different cell types that were generated to link regulatory single nucleotide polymorphisms (SNPs) to genes from Baur et al.15 The cell types represent major cell and tissue types studied by the NIH Roadmap Epigenomics Project.17 To create these networks we obtained the protein-protein interactions from the STRING database,18 RRID:SCR_005223. The protein-protein network is not cell type specific and combined it with cell type-specific TF-gene regulatory interactions. The regulatory interactions were defined in a cell-type specific manner and included both promoter-proximal and distal regulatory interactions. To create the cell-type specific regulatory networks, we downloaded Roadmap DNase I data from NIH Roadmap Epigenomics Project for each cell type and sequence-specific motifs for transcription factors from JASPAR,19 RRID:SCR_003030. Accessible motif instances were identified by intersecting motif instances with DNase I peaks on gene promoters and distal regions that exhibited long-range interactions with genes.15 The proximal TF-target regulatory network captures interactions between TFs and gene promoters exhibiting a significant and accessible motif instance. The distal regulatory network captures the interactions between TFs and genes, where the TF’s motif instance is on a region that interacts with the gene. The proximal and distal regulatory networks and the protein-protein interaction networks were merged by concatenating edges into one unweighted network per cell type. The statistics of these networks are described in Table 3.
Density is calculated by dividing the maximum number of edges in a full network by the number of edges. The values presented in the table are in mean (std) format.
Phenotype-specific diffused networks
We used the cell-type specific networks to identify parts of the network that were most affected by SNPs identified in different genome-wide association studies (GWAS,15). We refer to these as phenotype-specific networks. Briefly, for each of the 55 cell types, we scored genes in each network based on their propensity to interact with a non-coding SNP as assessed by the strength of the interaction between the region harboring a SNP and a gene promoter. The higher the score, the greater the strength of interaction between a gene and a region with the SNP. After assigning the node scores in the gene network, a two-step diffusion process was applied to first obtain scores for all other genes in the network using a regularized Laplacian kernel,20 followed by a second diffusion using a heat kernel to diffuse the signal among all genes. The output is a weighted adjacency matrix format where diagonal elements are node scores and off-diagonal elements are diffused edge weights. Since each cell type has a different network, we end up with 55 different nearly fully connected, weighted networks for each phenotype. We created these networks for SNPs from two phenotypes: Autism Spectrum Disorder (ASD) and Coronary Artery Disease (CAD). The statistics of these networks are described in Table 3.
Graph Laplacian embedding
The Graph Laplacian is a key operator for a given graph that has a large number of applications ranging from spectral clustering11 to using the Laplacian to smooth clusters on the graph.21–23 The normalized Graph Laplacian can be used to produce node embeddings as well and is computed as:
Here is the identity matrix and is the degree matrix obtained by summing the edge weights associated with each node and adding the average degree to the diagonal of the degree matrix. The addition of the average degree transformation was shown to be useful for real-world graphs with non-uniform degree distributions.24 Next, we compute eigenvectors associated with the smallest eigenvalues of the normalized Graph Laplacian matrix. Each node is then represented by corresponding entries in these eigenvectors to produce a -dimensional embedding of the node.
DeepWalk
The DeepWalk12 algorithm is based on random walks, which uses the skip-gram model25 to learn a node representation. We note that the authors of DeepWalk refer to this algorithm as a deep learning algorithm; however, we use the classification provided by Hamilton et al.,6 which considers DeepWalk as a shallow representation learning algorithm. In DeepWalk, each node’s representation is obtained from a set of random walks of a fixed window size on the graph starting from that node. For each node , the algorithm tries to find the embedding of node that maximizes the log-likelihood of neighbors observed in the random walk. This yields the optimization objective function that maximizes , where , the embedding of node and is a neighbor node. Naively optimizing this objective is computationally prohibitive, so DeepWalk uses a hierarchical softmax function where the neighbor nodes are stored as leaves on a binary tree and the above probability can be calculated by the product of the probability of observing nodes on the path from the root ():
As Deepwalk accepts unweighted graphs as input, we converted the fully connected weighted graph to an unweighted graph by keeping edges with the highest weights. Briefly, we computed the mean value of all edge weights in a network and used it as a threshold to remove the edges with weights lower than the threshold. Edges that remained in the graph were set to a weight of 1. After this step the networks for Autism Spectrum Disorder, the average node number reduces from 1358.45 to 1352.91 and the average edge number in the graphs reduces from 1,846,514.09 to 167,384.27 (approximately 1/10 of the edges in the original weighted networks), Table 3.
node2vec
Similar to the DeepWalk algorithm, node2vec13 also uses random walks and the skip-gram model on the graph to obtain node embeddings. However, in node2vec, biased random walks are used to better capture the node’s neighborhood information and are controlled by two parameters, and . The parameter controls the probability of revisiting a node right after moving away from it, while specifies the probability of visiting a node’s 1-hop neighbors.6 These parameters can be set to make the random walk behave more like a Breadth First Search algorithm (high ) versus Depth First Search (DFS, low ). As in DeepWalk, the embedding of a node is learned based on the probability of the neighbor set. This probability distribution is estimated efficiently by making the assumption that the probability of observing one node in a neighborhood is independent of another one and that a node and its neighbor nodes should have a symmetric effect on each other’s feature space. Let denote the set of neighbors of and denote the embedding of node . The independence assumption allows the probability of the neighbor set to be written as:
The probability of node pairs is defined as a softmax function. We define the likelihood of node pairs as a softmax function of:
node2vec implements negative sampling strategies when optimizing the likelihood function and updating the node’s embedding features. This improves computation efficiency and becomes more scalable compared to the DeepWalk algorithm. We explored a range of and values to explore neighborhoods using a more BFS or DFS algorithm. In addition to the default setting of , we also applied , with values . In order to facilitate the comparison between various settings of and parameters, we used the default embedding dimension proposed in the algorithm implementation. The difference in performance under various and values is minor across all values of , the number of clusters (Underlying data: Supplementary Figure 126,27). However, under the silhouette index, the default setting () always achieves top performance except when . For consistency with DeepWalk, we set the embedding space dimension to be (Table 4).
GraphSAGE
The GraphSAGE framework generalizes an inductive Graph Convolutional Network (GCN) framework and estimates a function with learnable parameters that can generate node embeddings based on the node attributes and the attributes of its neighboring nodes.14 The function in turn is a deep neural network where each layer makes use of aggregation and propagation of information from a node’s neighborhood. Given a learned function, GraphSAGE uses a forward algorithm to generate node embeddings, that consist of message passing and aggregation functions. In the message-passing step, each node retrieves its neighboring nodes’ current embedding and passes it through an aggregation function. This is denoted as
The hyperparameters of GraphSAGE include the number of hidden layers and the dimension of each layer. We set the number of layers to 2 as recommended by6 and constrain the number of units in both the hidden and output layers to be (Table 4). The objective function is optimized with batch gradient descent with a batch size of 256 and step size of 0.005. We also implement the negative sampling strategy by uniformly randomly selecting a target node to create an equal amount of negative links compared to positive ones. We use the mean aggregator function in the message passing step. As in the case of Deepwalk, we gave GraphSAGE unweighted versions of the phenotype-specific networks.
Variational graph auto-encoders (VGAE)
Graph auto-encoders (GAE)7 are commonly used unsupervised graph representation learning algorithms. They can be implemented either with or without node features. GAEs typically contain an encoder, which is often a Graph Convolutional Network (GCN)8 and a decoder, which uses inner products to predict edge presence. The loss function is based on the difference between the original adjacency matrix and the reconstructed one. Variational graph auto-encoder (VGAE)7 is an extension of GAEs that uses variational inference of model features. It assumes a prior distribution and encodes the node embeddings into a latent space via a GCN. In addition to the GAE loss function, its loss function minimizes the KL divergence between feature distributions and the standard normal distribution.
When training the VGAE model, we set the number of layers to two so that it is consistent with GraphSAGE and the input node feature matrix as an identity matrix. The hidden layers and embedding space dimensions are also set to be (Table 4). The size of the embedding space does not significantly impact VGAE’s performance as assessed on the cell-type specific networks (Underlying data: Supplementary Figure 226,27). Since the encoder of VGAE, GCN, is also based on the aggregation of node embeddings from their neighbors, under similar reasoning as GraphSAGE, VGAE was applied to unweighted versions of the phenotype-specific graphs.
To define cell clusters we applied the K-means clustering algorithm to the node embeddings obtained from each GRL algorithm. We set the number of clusters, , to vary from 10 to 100 with a step size of 10. To uniformly compare across algorithms, we had to pick a single that was optimal for all algorithms. To this end, we obtained the value for each method on each cell line that achieves the highest value based on the clustering evaluation metrics defined below. Then we select the mode value among all values to be and use the clustering results from this to compare across algorithms on different real and simulated networks. The optimal values for the simulated dataset was , for the cell type-specific networks was , and was for both phenotype-specific networks. For selected cell lines, we additionally visualized the modularity and silhouette scores for each algorithm as a function of (Underlying data: Supplementary Figures: 3–826,27). These values for individual cell lines are provided in Underlying data: Supplementary Tables 1–6.26,27
For the synthetic networks, we used three metrics to evaluate the quality of our clusters from each of the learned embeddings. Two of these, Modularity and Silhouette Index do not require any additional information to evaluate the quality of clusters. The third metric is Adjusted Mutual Information (AMI), which is used when there are ground truth labels for each cluster. AMI and Silhouette Index are widely used to assess the results from any clustering algorithm. Modularity is used specifically for graph clustering algorithms. All three metrics were used for the simulated networks, while SI and Modularity were used for the real networks.
Modularity
Modularity, proposed by Newman and Girvan,28 is the most widely used evaluation metric for the assessment on detected community structure. It represents the difference between the fraction of edges within one community and the fraction of edges in an equivalent network whose edges are randomly connected. Let denote the cluster , denotes the nodes in cluster , and denotes the set of edges. Modularity is defined as follows:
Here captures the proportion of edges within nodes of cluster and is the expected number of edges for members in cluster . The modularity score ranges from -1 to 1, with more positive values indicating greater community structure.
Silhouette Index
Silhouette Index29 measures how close a node is to its own cluster compared to its closest neighbor cluster and scores each node based on this schema. Each node’s overall average silhouette score is treated as a measurement of cluster quality. It is defined as:
Adjusted Mutual Information Index (AMI)
The mutual information (MI) score is a similarity measurement between two sets of clusterings, and defined as:
To interpret the clusters identified on the cell-type and phenotype-specific networks, we used GO enrichment analysis (Release 106).30–32 GO enrichment analysis was done using a Hypergeometric test with FDR correction. We set the threshold of q-value to be to define enriched terms. As GO enrichment can provide a large number of overlapping terms, we developed additional summary metrics to evaluate the GO enrichment: (a) the number of clusters enriched with any term, (b) the number of terms enriched in any cluster, (c) the distribution of the number of clusters a term is enriched in. This analysis was focused on the =50 results to obtain the greatest resolution of the clusters, while being optimal for most methods. Finally, we used terms that were deemed specific to a cell line (defined as enriched in a small number of clusters), to bicluster the cell lines and the terms using Non-negative matrix factorization (NMF).33 We first filtered out the GO terms that are enriched in more than 20% of the cell lines and created a GO term count matrix, where the row of the matrix is a GO term and the column represents the 55 cell lines. The entry of the matrix is the number of times a term is enriched in the corresponding cell line (maxed at the number of clusters). This matrix was factorized with NMF to group GO terms into distinct term categories and cell lines into different categories with 10 factors. This enabled us to associate groups of terms with groups of cell lines. We reordered the GO terms and cell lines based on the group assignments and visualized the count matrix as heatmaps (Underlying data: Supplementary Figure 10 to Supplementary Figure 1226,27). The code for doing the NMF-based ontology summarization is available at.26
We compared our three shallow (DeepWalk, node2vec, Graph Laplacian) and two deep (GraphSAGE, VGAE) GRL methods on simulated networks with known ground truth and real cell type-specific molecular networks.
We first evaluated the different methods on the synthetic graphs from the LFR benchmark using three metrics, Modularity, AMI and Silhouette Index. The synthetic graphs were generated by setting the mixing parameter, which controls the connectivity between clusters, to 0.1 or 0.5 (Methods). When , 10 percent of edges connected to each node link to another node in a different cluster and the remaining edges stay in the node’s cluster. Thus is easier to cluster compared to which has more inter-cluster edges. For , Graph Laplacian and node2vec embeddings performed overall the best for both modularity index (Figure 2A), followed closely by VGAE. VGAE achieved the highest score under silhouette index and AMI (Figure 2B & 2C). GraphSAGE outperformed others when evaluated by the modularity score, but had a relatively low score when considering the other metrics. This suggests that GraphSAGE is able to recover community structure better than other methods, even though the low dimensional embeddings of nodes within a cluster may not be as coherent as the other methods. These results also demonstrate the utility of assessing results using multiple metrics which can provide complementary information (e.g., modularity for GraphSAGE results). Deepwalk had the worst performance compared to other methods. Overall, VGAE, node2vec, and Graph Laplacian embedding had higher performance compared to GraphSAGE and Deepwalk.
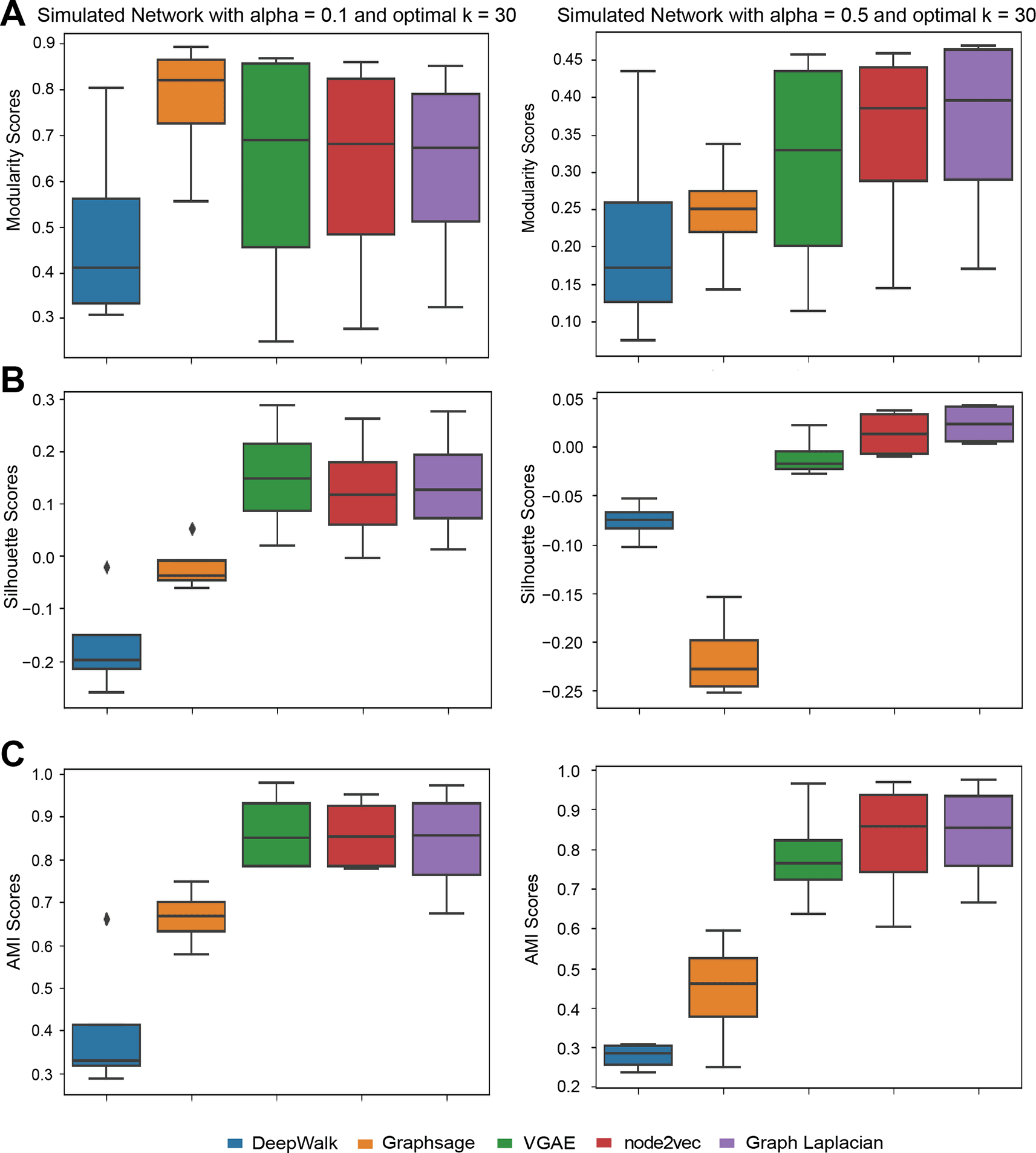
(A) The modularity scores with alpha = 0.1 and 0.5. (B) The Silhouette Index scores with alpha = 0.1 and 0.5. (C) The AMI scores with alpha = 0.1 and 0.5. Each algorithm is applied to simulated networks with the node degree [10, 25, 50, 100] and their scores form distributions that are summarized into box plots. For each data set, the optimal is chosen based on the overall performance of all algorithms.
For , which is more challenging from a clustering point of view, GraphSAGE and VGAE significantly dropped in performance under all three metrics. However, node2vec and Graph Laplacian were still robust for these graphs. GraphSAGE and Deepwalk were the two poorest-performing methods when aggregating the different metrics.
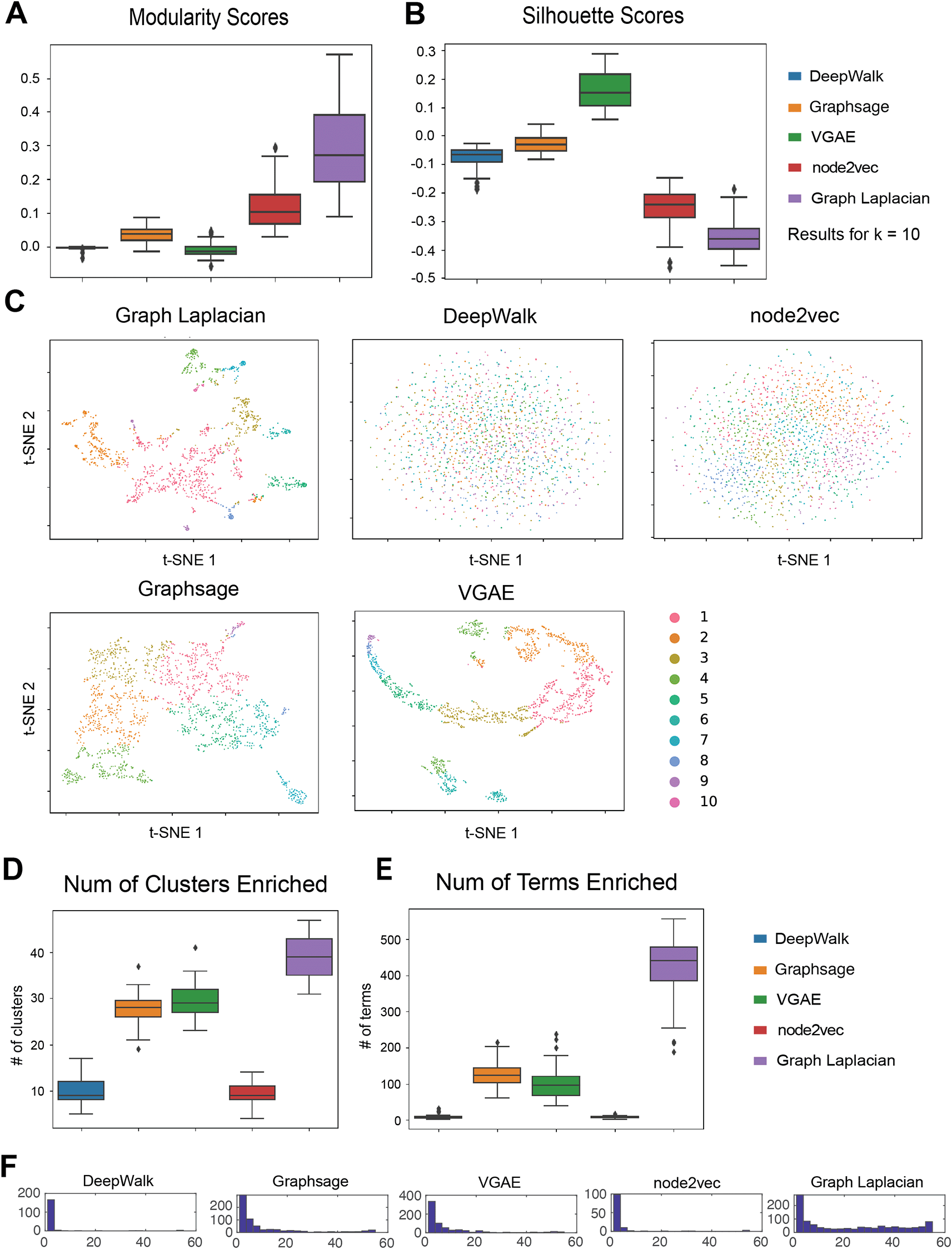
We next evaluated our five GRL methods for their ability to retrieve gene modules from 55 cell type-specific networks (Methods). These cell types corresponded to diverse cell lines and tissue types that were studied by the ENCODE ROADMAP consortium project.17 We first evaluated the quality of the modules based on Silhouette Index and Modularity, which did not require ground truth labels. The evaluation metrics were computed for each (number of clusters), to determine the best number of clusters for each algorithm for each cell line (Underlying data: Supplementary Table 126,27). To compare across algorithms, we used as this was the best across all cell lines and methods when using either modularity or SI. Furthermore, the choice of did not substantially affect the relative performance of algorithms (Underlying data: Supplementary Figure 326,27). Based on both modularity and SI for , DeepWalk performed the best, followed by Graph Laplacian (Figure 3A & 3B). Node2vec had a better performance under the modularity index but achieved a worse silhouette score. The deep GRL method VGAE had a better SI score than node2vec, but was outperformed on modularity by node2vec. GraphSAGE had the worst performance using both metrics. Overall, DeepWalk and Graph Laplacian embedding had a consistently high performance using both metrics. Deep GRL methods had poor performance for Modularity, but we saw better silhouette scores for the VGAE algorithm.
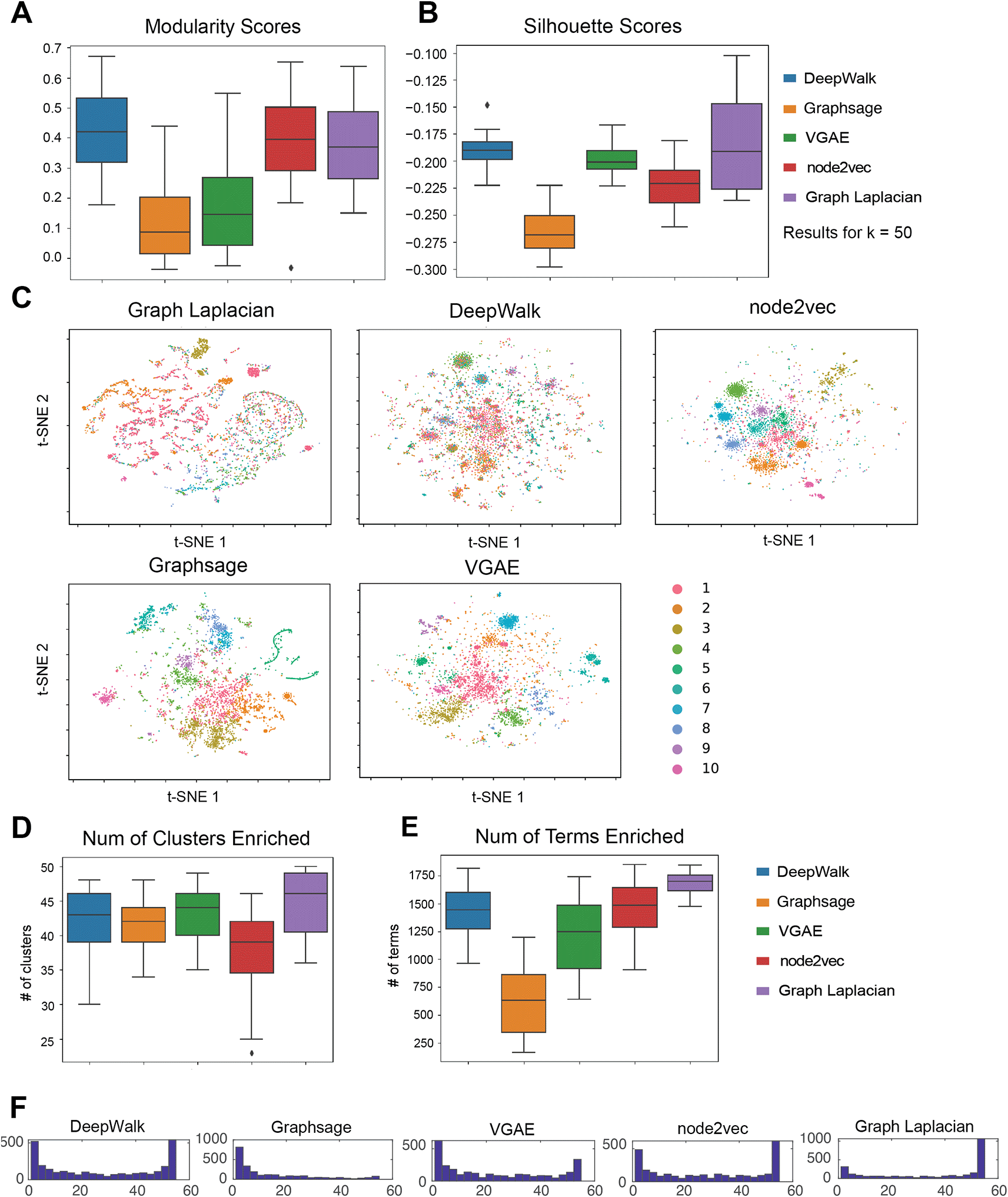
(A) The distribution of modularity across 55 cell-type specific networks. Each algorithm is applied to 55 cell line networks and their scores form distributions that are summarized into box plots. For each data set, the optimal is chosen based on the overall performance of all algorithms. (B) The distribution of Silhouette Index scores across the 55 cell type-specific networks. As in Modularity, we picked the that was best for each cell line. In general, Modularity and Silhouette Index picked a similar . (C) tSNE visualizations of the embeddings learned by each method. The colors correspond to the different clusters. The colors are matched across methods so that the largest cluster is shown as cluster ID 1, followed by cluster 2, etc. (D) Number of clusters enriched with a GO term in 55 cell line networks. (E) Number of terms enriched in any cluster in 55 cell line networks. (F) Distribution of the number of cell lines a term is enriched in any cluster.
To further examine the inferred clusters, we used tSNE34 to visualize the node embedding of genes for each method in a two-dimensional space (Figure 3C) and color the nodes based on their cluster membership. To be consistent with the above analysis, we selected cluster assignments with = 50 and plotted the 10 largest clusters. We selected the heart cell line since it had the highest Modularity across methods. For all methods, there was a clear visual separation between the members of each cluster (different colors). The tSNE visualization also showed that the Graph Laplacian embedding tended to identify less uniform-sized clusters, with two large clusters (2922-1448 genes) in comparison to the other methods.
We next interpreted the cluster assignments by performing GO enrichment analysis. Across 55 cell-type specific networks, we counted the number of clusters (out of a total of 50) that are enriched for a GO process term (Figure 3D). Across all methods, the majority of the clusters were enriched, with Graph Laplacian producing the largest number of enriched clusters across cell lines followed by DeepWalk and VGAE. Node2vec had the lowest average number of enriched clusters. This indicates that Graph Laplacian, DeepWalk and VGAE obtained the most biologically meaningful clusters for these networks. We next counted the total number of terms enriched in any cluster for each of the methods (Figure 3E). Across the 55 cell lines, we found that Graph Laplacian had the greatest number of terms on average, which was followed by node2vec and DeepWalk. Here, GraphSAGE had significantly fewer terms compared to any of the methods. We aimed to assess the overall specificity of the clusters for the GO terms by examining the distribution of the number of clusters a term is enriched in. We expect this distribution to be bimodal, with some general terms to be enriched in many clusters, while more specialized terms would be enriched in fewer clusters (Figure 3F). Here DeepWalk, VGAE and node2vec exhibited the clearest bimodal distributions. Graph Laplacian embedding tended to identify more generic terms (inferred in many clusters) and relatively fewer cluster-specific terms compared to other methods. In contrast, GraphSAGE had the opposite behavior with most terms enriched in a smaller number of clusters. In parallel, we examined the distribution of the number of cell lines a term was enriched in each of the GRL methods. Most methods with the exception of GraphSAGE exhibited a bimodal distribution, with most terms either being enriched in many of the cell lines (50) or few cell lines (4). Graph Laplacian was unique in that most of the terms were shared across cell lines. Finally, we used the specific GO terms for each method (defined by biclustering cell lines and the enriched GO terms using non-negative matrix factorization (Underlying data: Supplementary Figure 10,26,27 Methods). Both DeepWalk and VGAE grouped the fetal cells separately, providing more skewed cell line clusters, while the other methods provided more uniform groups of cell lines and their corresponding terms. The association of the GO terms with cell line clusters was consistent with the role of the cell lines for these methods. For example, in Graph Laplacian, we identified the immune cell lines associated with immune function, and developmental processes were associated with the embryonic stem cell lines. Taken together, these results show that shallow representation learning methods are able to recover biologically meaningful groupings of cell lines.
Network-based approaches offer a powerful framework for interpreting sequence variants identified from GWAS by identifying pathways that might be jointly perturbed by a set of sequence variants.35,36 A common theme of these approaches is to map a set of sequence variants onto a network and interpret these variants based on subnetworks connected by these variants. As another application of GRL, we examined how the embedding affects the results for network-based GWAS interpretation. To this end, we considered two phenotypes, Autism Spectrum Disorder (ASD) and Coronary Artery Disease (CAD), which are among the most well-studied phenotypes in the NHGRI catalog.37 We focused specifically on non-coding variants, which are more challenging to interpret as they are often far away from genes. We used heat diffusion kernel to first construct weighted graphs for each phenotype (Methods), and then applied each GRL method to obtain an embedding followed by k-means clustering (Figure 4, Figure 5). Because GraphSAGE and DeepWalk require unweighted graphs, we first removed edges with weights less than the average edge weight per cell line and then gave the remaining edges as an unweighted graph as input to GraphSAGE and DeepWalk. For the VGAE algorithm, the encoder was based on Graph Convolutional Network that learns each node’s embeddings from its neighborhood. Since both ASD and CAD phenotype-specific networks were nearly fully-connected, this can produce noisy embeddings. Thus, we applied VGAE on the unweighted graphs as well.
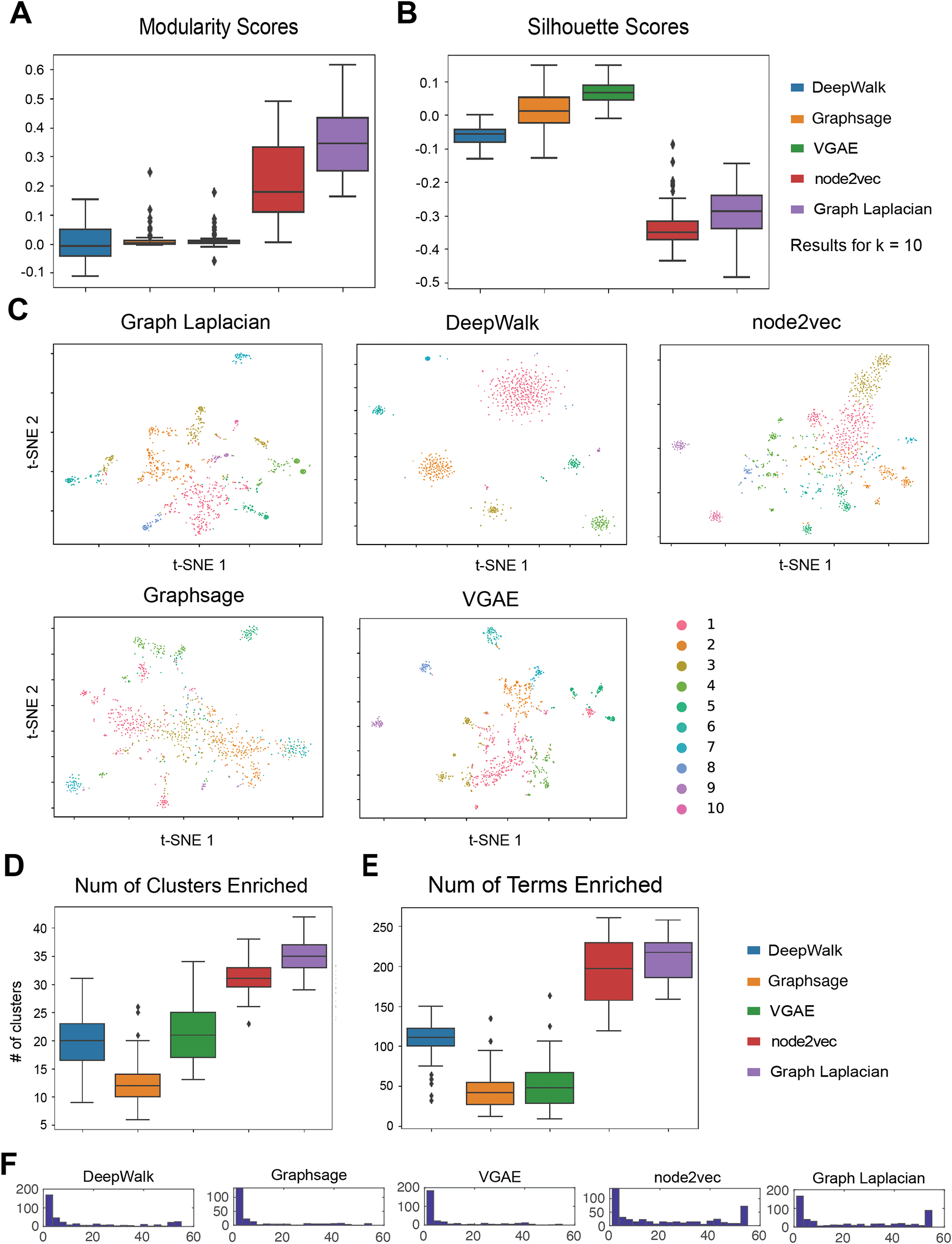
(A) The distribution of modularity across 55 cell line networks obtained with ASD. Each algorithm is applied to 55 cell line networks and the scores are summarized in a box plot. Shown are the results for , which was chosen as the optimal based on the overall performance of all algorithms. (B) The distribution of silhouette scores across the 55 networks when using clusters. (C) tSNE visualizations of the embeddings learned by each method. The colors correspond to the different clusters. The order of cluster numbers matches the decreasing order of cluster sizes. (D) Number of clusters enriched with a GO term in 55 cell line networks. (E) Number of terms enriched in any cluster of the 55 cell line networks. (F) Distribution of the number of cell lines a term is enriched in any cluster.
(A) The distribution of modularity across 55 cell line networks obtained with CAD. Each algorithm is applied to 55 cell line networks and the scores are summarized in a box plot. Shown are the results for , which was chosen as the optimal based on the overall performance of all algorithms. (B) The distribution of silhouette scores across the 55 CAD phenotype-specific networks when using clusters. (C) tSNE visualizations of the embeddings learned by each method. The colors correspond to different clusters. The order of cluster numbers matches the decreasing order of cluster sizes. (D) Number of clusters enriched with a GO term in 55 cell line networks. (E) Number of terms enriched in any cluster in 55 cell line networks. (F) Distribution of the number of cell lines a term is enriched in any cluster.
As in the analysis of the cell-type specific networks, we chose the optimal value based on the mode of the Modularity and Silhouette Index values across 55 cell lines and all 5 methods (Underlying data: Supplementary Table 3–6,26,27). The optimal under these two metrics was 10. Based on the modularity metric, the top-performing method was Graph Laplacian followed by node2vec for the ASD phenotype network (Figure 4A). However, with the Silhouette Index, Graph Laplacian and node2vec had the worst performance (Figure 4B). Here, VGAE exhibited the best performance followed by GraphSAGE and node2vec. VGAE, GraphSAGE and DeepWalk accepted undirected weighted graphs and the opposite behavior of these algorithms for modularity and Silhouette Index could be due to that. These results demonstrate the benefit of deep learning frameworks for embedding graphs for GWAS analysis. We also visualized the clusters obtained for ASD phenotype networks in the heart cell line with t-SNE plots (Figure 4C). For all algorithms, the clustering structure was visually discernible with genes in the same cluster being positioned close together.
We next evaluated the different GRL methods based on GO enrichment of the inferred clusters. Here we considered to have a sufficient granularity of the clusters to capture specific biological processes. We examined the distribution of the number of clusters enriched with a GO process as well as the number of GO process terms that were enriched across the 55 cell lines. Graph Laplacian embedding produced the highest number of clusters enriched with biological functions, followed by node2vec (Figure 4D). DeepWalk and VGAE had a similar average number of clusters enriched and GraphSAGE had the fewest. GraphLaplacian and node2vec were equally good when considering the number of terms followed by DeepWalk (Figure 4E). Both GraphSAGE and VGAE had the fewest number of GO terms. Finally, we examined the distribution of the number of clusters for each term (Figure 4F). Only Graph Laplacian and node2vec exhibited a bimodal distribution, meaning that they can discover both generic and specific term GO terms. As we did previously, we selected the most specific terms (defined by their enrichment in any cluster of 20% of the cell lines) and applied NMF to find cell line and term clusters (Underlying data: Supplementary Figure 1126,27). Graph Laplacian and node2vec exhibited the clearest block diagonal structure with groups of cell lines associated with groups of GO terms. Several of these groupings were biologically meaningful, for example, the association of muscle and cardiac functions with a cell line cluster with heart (Graph Laplacian) and immune response associated with cd14 primary cells (node2vec).
We performed a similar analysis for the CAD phenotype network as well (Figure 5). We observed similar trends for the relative algorithm performance when using Modularity and Silhouette Index, with methods using undirected graphs exhibiting a higher Silhouette Index compared to methods using the weighted graphs (Graph Laplacian and node2vec) and an opposite trend when using Modularity (Figure 5A & 5B). Visualization of the tSNE plots for the heart cell line showed that while Graph Laplacian, GraphSAGE and VGAE retrieved layouts that were consistent with the clustering, DeepWalk and node2vec visualization were noisy (Figure 5C). A similar noisy layout was observed for other cell lines as well suggesting that the CAD networks were noisier than the ASD networks (Underlying data: Supplementary Figure 926,27). Based on the number of enriched clusters and terms enriched, Graph Laplacian performed best, followed by VGAE and GraphSAGE (Figure 5D). The low enrichment of the DeepWalk and node2vec algorithms is consistent with the lack of clustering structure in the tSNE results. The histogram of term specificity exhibited a bimodal distribution only with the GraphLaplacian embedding (Figure 5E); however, all three methods (Graph Laplacian, VGAE and GraphSAGE) produced well-separated cell line and biological process groups (Underlying data: Supplementary Figure 1226,27).
Overall, our comparison of the GRL methods for the GWAS interpretation task showed that Graph Laplacian provided the most robust embedding under all metrics. Furthermore, the performance of the methods depended upon the metric and phenotype and deep GRL methods were less prone to noisy diffusion-based graphs compared to random-walk based methods (node2vec, DeepWalk).
Graph representation learning (GRL) has emerged as a powerful paradigm for applying machine learning algorithms on graphs. More recent deep graph representation learning algorithms have shown improved performance on numerous graph machine learning problems such as node classification and link prediction. Given the modular organization of molecular networks such as protein-protein and regulatory networks, a natural question is whether deep GRL methods have an advantage for detecting modules or clusters on graphs over shallow GRL methods. In this paper, we performed a comprehensive benchmarking study on synthetic networks and real molecular networks capturing protein-protein and regulatory interactions. On real networks, we considered two types of applications: detection of gene modules from cell type-specific networks and detection of candidate pathways affected by single nucleotide polymorphisms identified in GWAS. We observed some benefits of deep GRL algorithms on simulated networks as well as in situations where the graph may be noisy.
Our experiments on simulated networks were helpful because of the availability of ground truth and a controlled experimental setting. In particular, the mixing parameter of 0.1, which results in sharper boundaries across clusters, all but DeepWalk perform well using Modularity. However, for a more challenging cluster structure, GraphSAGE dropped in performance. Between VGAE and GraphSAGE, which are both deep methods, VGAE was consistently better. It is likely that GraphSAGE is better for graphs where node attributes provide complementary information to the graph structure. When the networks are sparse, VGAE achieved equally good performance as node2vec and Graph Laplacian embeddings. However, shallow methods consistently outperformed deep learning frameworks for denser graphs where modules are harder to detect. Our experiments showed that deep GRL methods such as VGAE can outperform other algorithms but this depends upon the structure of the graph and the specific metric.
On real molecular networks, the relative performance of the algorithms depended upon the task. When defining gene modules across 55 cell line/type-specific networks, shallow GRL methods consistently outperformed both VGAE and GraphSAGE when using Modularity. However, when using the Silhouette Index, VGAE was comparable to shallow methods but GraphSAGE remained worse. Interestingly, despite low silhouette scores, all methods generated well-clustered tSNE visualizations and identified clusters that were enriched for gene ontology processes. When considering the task of GWAS interpretation, we found both GraphSAGE and VGAE and the shallow method, DeepWalk, to produce better silhouette scores, but this trend was reversed when using modularity. This observation was reproducible for the second phenotype as well. All three algorithms accepted filtered undirected graphs as input, while GraphLaplacian and node2vec accepted nearly fully connected weighted graphs. The pre-processing of graphs before providing them as input to graph embedding algorithms could be a major determinant of an algorithm’s performance. When compared to the ASD phenotype-specific graphs, we found DeepWalk and node2vec to produce noisy tSNE visualizations, which could be due to the greater noise in this graph and the susceptibility of random walk methods to noisy graphs. This was consistent with the relatively lower modularity score of node2vec compared to Graph Laplacian as well as a smaller number of enriched terms for these methods. Finally, across all network types, tasks and metrics, we found Graph Laplacian to provide consistently good embeddings, though it was not necessarily the best for some conditions. The only potential limitation of the Graph Laplacian is to infer unbalanced gene clusters for the cell-type specific networks. A direction of future work would be to consider alternative pre-processing of the graph or additional regularization to obtain more uniformly-sized clusters.
Our study compared select deep GRL and shallow GRL algorithms on graphs that did not have any node attributes. One direction of future work is to expand the type of graphs to those with node attributes and accordingly increase the class of algorithms to be compared. Another direction of future work is to detect dynamic gene modules, for which there are shallow38 and deep learning algorithms.39
In conclusion, our benchmarking study shows that the performance of different embedding methods depends on the metrics used as well as network characteristics, such as sparsity and edge weight distribution. Deep GRL algorithms are not necessarily advantageous over shallow methods for module detection and this depends upon the algorithm, the type of graph and the specific module-based task. Between the two deep GRL algorithms, we found the VGAE algorithm to offer superior performance. Comparing across different graph types and module metrics, Graph Laplacian provided consistently robust and high-quality embeddings suggesting that it could be a good baseline algorithm to compare for newly developed representation learning algorithms.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)