Keywords
PCR, RNA, Viruses, RT transcriptase, Moloney Murine Leukemia Virus enzyme
PCR, RNA, Viruses, RT transcriptase, Moloney Murine Leukemia Virus enzyme
Reverse transcriptase (RT), also known as directed DNA polymerase, is an enzyme encoded by the genetic material of retroviruses, which catalyzes the conversion of RNA to DNA in a process called reverse transcription.1 It allows retroviruses, such as Human Immunodeficiency Virus (HIV), to form duplicates and integrate into the host genome. Three distinct biochemical characteristics facilitate the enzymatic activity of RT: Ribonuclease H, RNA-dependent DNA polymerase, and DNA-dependent DNA polymerase.2 The RT structure is a ‘right hand’ characteristic of cellular DNA polymerases, having palm, thumb, and fingers subdomains, and also a spatially detached RNase H domain.3,3
RT characterized by good catalytic activity, low RNase H activity, and high temperature stability has been reported to be more acceptable for biotechnological applications.4 Two virus-derived genes have been used to produce recombinant RTs: avian myblastosis virus (AMV) and Moloney Murine Leukemia Virus (MMLV). Among all the known RTs, the most studied is the reverse transcriptase from the Moloney Murine Leukemia Virus (MMLV), which is a 75kiloDalton protein containing five domains, the palm, thumb, and fingers, making the polymerase, connection domain, and RNase H domain, and is more active in the monomeric state.5 Using a thermostable variant of RT is advantageous for reducing non-specific nucleic acid amplification and minimizing the influence of intricate secondary structures.6
MMLV possesses two active sites for viral RNA reverse transcription, both of which require Mg2+ or Mn2+ for optimal catalysis. Recombinant MMLV reverse transcriptase is preferred due to its thermostability as compared to its wild-type, as a result of the reduced RNase H activity of the RT.5 Additionally, its robust catalytic activity and high fidelity make RT the most widely used enzyme.7
Infectious illnesses, particularly those of viral origin, spread like wildfires, and are the leading cause of disruption in livelihood, trade, and the economy. Viruses such as Ebola, Lassa fever, Yellow Fever, Zika, Chikungunya, H1N1 influenza, Middle East Respiratory Syndrome (MERS), Severe Acute Respiratory Syndrome (SARS), and continuing COVID-19 are just a few of the epidemics that have hit the public in this era.8 Viruses that contain RNA have been found to be the primary etiological agents of severe and emergent pathogens in animals and humans,9 occupying for approximately 44% of all emerging infectious diseases in humans. The detection of these RNA viruses, such as SARS-CoV-2, requires the use of reverse transcriptase (RT) polymerase chain reaction (PCR), where the RNA is converted to DNA through a cDNA intermediate in PCR amplification.10
Reduction in the activity of RTs is a common occurrence in resource-limited countries, where cold chain storage is a major challenge because of the lack of electricity, interruption of electricity, or high cost of running diesel generators, especially in third-world countries.11 The aim of the present study was to generate and characterize a variant of the MMLV-RT Enzyme.
The bacterial strain used in this study was ECOS-BL21 (DE3)-competent E. coli cells obtained from Invitrogen. Genscript Biotech Corporation synthesized and integrated the MMLV-RT gene into the pET28a (-) plasmid, which also featured the isopropyl β-d-1-thiogalactopyranoside IPTG-inducible T7 promoter, Ori-pUC, and kanamycin antibiotic marker. The SARS-CoV2 samples used in the evaluation phase of this study were archived samples from the Kenyatta National Hospital Infectious Disease Unit from patients with SARS-CoV2 at KEMRI’s Innovation and Technology Transfer Division.
This study employed cross sectional study design.
The sample size for the SARS-CoV 2 specimens used in this study was determined using Fisher’s exact formula as described below:
Fisher’s formula for sample size calculation.
Z = normal standard deviation, normally set to 1.96, corresponding to 95% C.I.
The prevalence of SARS CoV2 in Kenya is 5.6% (0.056).12
ε = Margin of error, normally set at 5% (0.05)
Therefore, a minimum of 81 samples were expected to be used for enzyme evaluation. However, this study utilized 129 SARS-CoV 2 samples for the performance evaluation of the expressed in-house recombinant MMLV-RT Enzyme.
The six latest sequences encoding thermostable Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV-RT) were identified and retrieved from the NCBI website (https://www.ncbi.nlm.nih.gov/). The sequences were aligned using multiple sequence comparisons by log-expectation (MUSCLE) (https://www.ebi.ac.uk/Tools/msa/muscle/). Consensus sequence construction was performed using EMBOS Cons (https://www.ebi.ac.uk/Tools/msa/emboss_cons). The obtained consensus sequence was aligned against six sequences.
The consensus sequence obtained was codon-optimized using GenSmart™ Codon Optimization https://www.genscript.com (, Genscript Biotech Corporation). To obtain a more optimal codon sequence, the codon-optimized gene sequence was analyzed using the GenScript Rare Codon Analysis tool (GenRCA). The GenRCA tool calculated the Codon Adaptation Index (CAI) and percentage GC content of the generated genetic sequence encoding MMLV-RT.
Both Histidine tag and enterokinase cleavage sites were added to the MMLV-RT-encoding gene before cloning. After verification of the codon-optimized MMLV-RT gene, it was synthesized and cloned into the pET28a (−) vector (Genscript Biotech Corporation).
The MMLV protein in the pET28a expression vector was transformed in ECOS-BL21 (DE3) (Invitrogen) competent cells as previously described.13,14 Briefly, 10 μL of ECOS-BL21 (DE3) competent cells (on ice) was dispensed into ten 2 mL Eppendorf tubes labeled BL21-DE3. Then, 1 μL of pET28a expression vector carrying MMLV-RT recombinant DNA was added to each tube, mixed by tapping, and left to disperse evenly for 5 min on ice. The cells were heat-shocked at 12oC °C for 15 s in a water bath at 37oC °C. The cells were then placed on ice for 2 minutes. The culture was shaken for 1 h to allow for kanamycin-resistant growth. Kanamycin was then added at a final concentration of 100 μg/mL and cultured in 10 mL LB broth at 230 rpm overnight while shaking in an incubator shaker (INNOVA 42). The turbidity of the LB medium was assessed to confirm the growth of the cultured cells. All small cultures were then transferred to a 500mL LB-kanamycin media and incubated overnight at 37 °C with shaking at 230 rpm for approximately 1 h and 45 min. Cell growth density was then measured using a DEN-1B McFarland densitometer (Waken Teck. Co., Ltd.) with the aim of attaining a range of 2.5-3.0 density.
After transformation, the cells were grown in 10 ml LB kanamycin overnight at 37 °C with shaking at 230 rpm. Upon attaining a cell growth density of 3.98 cell/mL, protein expression was induced by the addition of 500 μL of 1 M IPTG (Isopropyl-β-d-1-thiogalactopyranoside) (Scientific, Inc.) and incubated at 37 °C with shaking at 230 rpm for 1 h. Thereafter, the density was measured to assess cell growth using a DEN-1B McFarland densitometer (Waken Teck. Co., Ltd.). The culture was then centrifuged at 3000 g for 20 min at 20 °C using an Eppendorf 5810R centrifuge. The MMLV-RT recombinant protein extract was obtained as an insoluble fraction that contained inclusion bodies. The pellet obtained was suspended in 10 mL Bugbuster (Novagen, United Sates) containing 1 μL benzonase and 1 μL lysozyme (Novagen, USA).
The MMLV-RT recombinant protein extract pellet was mixed with 10 mL of 1x Bugbuster through hard pipetting, followed by the addition of 1 μL of lysozyme. The resulting mixture was then slowly rotated at room temperature for 15 min. Then, 10 mL of 1/10 Bugbuster solution was added and mixed by vortexing for 1 min. The mixture was then centrifuged at 5000 × g for 15 min at 4 °C, and the resulting pellet was washed by suspending it in 20 mL of 1/10 Bugbuster solution and vortexing for 1 min. This washing step was repeated twice, and after the final washing step, the obtained pellet was suspended in 10 mL of 1/10 Bugbuster solution by agitation for 1 min.
The recombinant protein suspension obtained from inclusion bodies was centrifuged at 12 rpm for 15 min at 4 °C. The resulting inclusion body pellet was mixed with 10 mL solubilization buffer (0.3% N-LS/CAPS, pH 11) through hard pipetting for 1 min. The mixture was then rotated at room temperature for 15 min and spun at 12 rpm for 15 min at 4 °C, resulting in a soluble MMLV-RT recombinant protein extract that was ready for use in subsequent steps. To increase the concentration of MMLV-RT, protein was dialyzed in 1 liter of 0.1% N-LS/PBS (-), pH 7.6 at 4 °C for 2 h. The dialysis medium was then replaced with fresh medium and dialysis was repeated for another 2 h. This process was repeated, after which the medium was replaced with fresh dialysis medium and left to dialyze overnight at 4 °C.
To purify the MMLV-RT expressed protein, Immobilized Metal Affinity Chromatography (IMAC) was carried out using TALON-Accept resin (Code #635503, Takara Bio Inc.) with slight modifications to the manufacturer’s instructions. The His-accept resin was transferred into an empty column (Bio-Rad), washed with double-distilled water, and equilibrated using 4 mL of dialysis buffer. Next, 10 mL of soluble fraction supernatants containing MMLV-RT Enzyme was loaded onto the His-Accept resin, and the His flow-through (His-FT) solution was collected. The resin was washed with 4 mL of 1x Bugbuster and then washed twice with 10 mL of NPT-I10 solution. Finally, the MMLV-RT enzyme was eluted using NPT-I1300 elution buffer, and the His-elute solution was collected.
The Pierce BCA protein assay kit (Thermoscientific, USA) was used to determine
Quantity of pure MMLV-RT-expressed protein. Standard solutions and working reagents were prepared. Twenty-five microliters (25 μL) of each standard solution and His-Elute (MMLV-RT enzyme) was separately transferred into labeled tubes. Then, 500 μL of the working reagent was added to each tube, covered with aluminum foil, and incubated in a water bath at 37 °C for 30 min. The MMLV-RT recombinant protein was quantified using the BCA protein assay (562 nm) on a Thermo Scientific NanoDrop 2000c UV-Vis spectrophotometer (USA). Absorbance for the blank was used as background, and measurements were taken from low to high concentrations of the standard solutions, then for recombinantly produced MMLV-RT enzyme within 10 min.
Two gels were prepared to evaluate the quality and purity of MMLV-RT protein. One-dimensional SDS-PAGE was used to separate purified proteins on 10% gels. To determine the purity of specific proteins, one gel was stained with Coomassie Brilliant Blue, washed, de-stained using methanol, and imaged using an Azure 280 chemiluminescent imaging system. Another gel was used for western blotting to determine the quality of the purified proteins. The gel was transferred onto a polyvinylidene fluoride (PVDF) membrane using a Bio-Rad Trans-Blot system. The PVDF membrane was blocked, washed, and incubated with diluted MBL-anti-His-tag HRP direct-tagged antibodies. The membrane was washed and incubated with ECL solution before imaging using an Azure 280 chemiluminescent imaging system.
RNA was extracted from SARS-CoV 2 samples using the QIAmp RNA mini-extraction kit (Qiagen Inc., USA) according to the manufacturer’s instructions. Briefly, 560μL of lysis buffer (AVL) was mixed thoroughly with carrier RNA. 140μL of the sample was added and incubated for 10 min at room temperature. The mixture was transferred to a spin column after the addition of 560μL of absolute ethanol. The spin column was spun twice at 8000 rpm for 1 min each. The contents were then washed with 500 μl both AW1 and AW2. The RNA was then eluted from the column using 60 μl AVE buffer and stored at -30°C.
The status of the 129 SARS-CoV 2 samples to be used in the in-house enzyme testing was verified using real-time RT-PCR. The qPCR Master Mix was prepared as follows: 2X GoTaq® qPCR Master Mix (Cat. # M7122, Promega Corporation, Madison, USA); 10 μl of 2X GoTaq® Green Master Mix, 0.5 μl NIID_2019_nCoV_N_F1 (10 μM), 0.5 μl NIID_2019_nCoV_N_R1 (10 μM), 0.5 μl NIID_2019_nCoV_N_Prb (10 μM) and 3.5 μl of DEPC treated Water. The Template RNA (5 μL Template RNA was added to a total volume of 20 μL. The primers used in this verification step is as shown in Table 1 below.
| Primer name | Primer sequence25 |
|---|---|
| NIID_2019_nCoV_N_F1 | 5’-AAATTTTGGGGACCAGGAAC-3’ |
| NIID_2019_nCoV_N_R1 | 5’-TGGCAGCTGTGTAGGTCAAC-3’ |
| NIID_2019_nCoV_N_Prb | 5’-FAM-ATGTCGCGCATTGGCATTGGCATGGA-BHQ-3’ |
The cycling conditions were as follows: reverse transcription at 50 °C for 15 min, initial denaturation at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 3 s and annealing and extension at 60 °C for 60 s. The results were viewed and interpreted as either RT-PCR positive or negative, depending on the CT values.
The activity of the purified MMLV-RT was determined by reverse transcribing the extracted mRNAs from SARS-CoV 2 positive and negative samples. This was performed using SuperScript III Reverse Transcriptase and in-house RT. The following components were combined in a nuclease-free microcentrifuge tube: 70 ng random primers, 10 pg total RNA, and 1 μL 10 mM dNTP mix. The volume was adjusted to 13 μL by using sterile distilled water. The mixture was heated at 65°C for 5 min and then cooled on ice for 1 min. After brief centrifugation, 4 μl of 5X First-Strand Buffer, 1 μl of 0.1 M DTT, 1 μL of RNase OUT ™ Recombinant RNase Inhibitor (Cat. no. 10777-019, 40 units/μL), to which either 1 μL of SuperScript™ III RT (200 units/μL) or 1.5 μl of the in-house purified reverse transcriptase was added. The contents were gently mixed by pipetting, followed by incubation at 25°C for 5 min and then at 50°C for 60 min. The reaction was terminated by heating the mixture at 70°C for 15 min.
PCR was performed using The Prime Script two-step RT-PCR Kit (Cat. No. RR014B Takara Bio Inc.). Each PCR tube contained a total volume of 25 μL reaction mixture, with 5μl of cDNA template, 10 μl of 2X PCR Master mix (Primescript, Takara Bio Inc.), 1 μL each of a 10 μM concentration of RdRp forward primer and RdRp reverse primer, and 8 μL of nuclease-free water. The mixture was then loaded into a thermocycler (ABI Systems). The amplification PCR profile was set at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 60 s, and extension at 72°C for 60 s. The final extension step was performed at 72°C for 5 min.
The primers shown in Table 2 were used for the amplification of cDNA using conventional PCR.
| Primer name & Size (bases) | Primer sequence |
|---|---|
| RdRp_F1 (23) | 5’-CAAGTGGGGTAAGGTAAGGCTAGACTTT-3’ |
| RdRp_R1 (22) | 5’-ACTTAGGATAATCCCAACCCAT-3’ |
For visualization, a 1.5% agarose gel was prepared using SafeRed (Applied Biological Materials Inc., British Columbia, Canada) in 1x TBE buffer, and 5 μL of the product was loaded onto the gel and run at 100 V for 45 min. Imaging was performed using an Azure 600 Gel Imaging System (Azure Biosystems Inc.).
Twenty Samples (20) were used to test the In-house RT against commercial RT. Out of the 20 samples, 15 were positive for SARS-CoV 2 and 5 were negative samples and one was a negative control. This was done in comparison with a commercial RT kit, namely superscript (Invitrogen).
To evaluate the In-house Reverse Transcriptase, 129 SARS-CoV 2 samples were used, of which 89 RT-PCR confirmed positive and 40 negative samples, and a negative control. The samples were initially subjected to total RNA extraction followed by cDNA construction using the developed recombinant In-house MMLV-RT enzyme. The resulting cDNA was amplified by conventional PCR under previously specified conditions. The outcomes were recorded in Microsoft Excel for analysis and compared to the status of the SARS-CoV-2 samples, as verified by RT-PCR. The sensitivity (Se), specificity (Sp), positive predictive value (PPV), negative predictive value (NPV), agreement measured by Cohen’s kappa value, and overall accuracy were computed. Receiver operating characteristics were utilized to generate the area under the curve, illustrating the diagnostic capability of the In-house MMLV-RT enzyme.
This study was approved by the Scientific Ethics Review Unit of the Kenya Medical Research Institute on June 13th 2021 (approval number: KEMRI/SERU/CBRD/22/4208.
Data analysis was performed using Microsoft Excel 2017 and GraphPad Prism Version 10. To evaluate the developed Recombinant In-house MMLV-RT enzyme activity, we assessed the sensitivity (Se) specificity (Sp) value (PPV) and negative predictive value (NPV) agreement measured by Cohen’s kappa value. Receiver operating characteristics were employed to generate the area under the curve, illustrating the diagnostic capability of In-house RT. In the computation of P-values, a two-sided Fisher’s exact test was conducted using a significance value of <0.05. The Wilson-Brown method was used to compare the confidence intervals of the developed enzyme to those of a standard commercial kit.
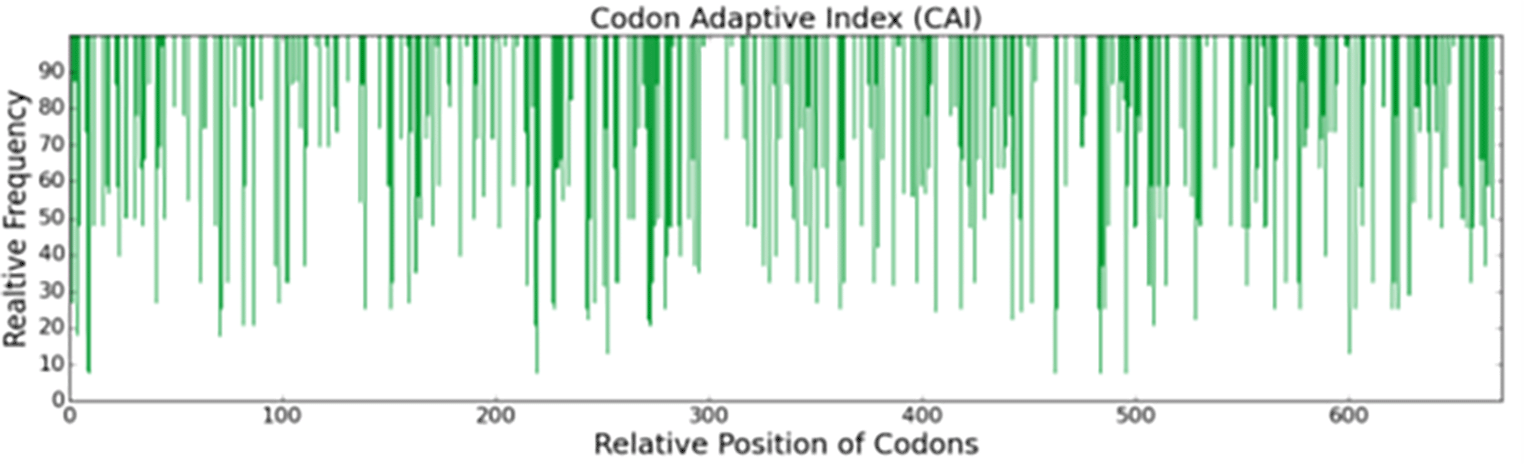
The consensus sequence derived from the six most recent sequences in the Genebank database, optimized for the expression of the recombinant His-MMLV-RT gene in E. coli (BL21) when codon optimized, exhibited a Codon Adaptation Index (CAI) of 0.72 (range 0-1), suggesting potential for expression. The sequence had an average GC content of 57%. (Table 3 and Figure 1).24
| Gene Name | Codon Adaptation Index (CAI) | Average %GC Content | Percentage GC Content at three Positions of Synonymous Codons | ||
|---|---|---|---|---|---|
| Position 1 | Position 2 | Position 3 | |||
| MMLV-RT Gene | 0.72 | 57 | 63 | 44 | 64 |
After the transformation process, the density (DEN-1B) of E. coli (BL21) cells was measured before and after protein expression. The density increased after the IPTG addition from 3.98 cells/mL to 6.98 cells/mL indicating successful protein expression (Table 4).24
The expressed protein was quantified using the Pierce BCA Protein Assay kit (Thermo Scientific) and found to be 0.367 mg/mL.
SDS-PAGE was used to verify the successful production of the in-house MMLV-RT. Coomassie Brilliant Blue analysis confirmed the purity of MMLV-RT, as shown in Figure 2.24
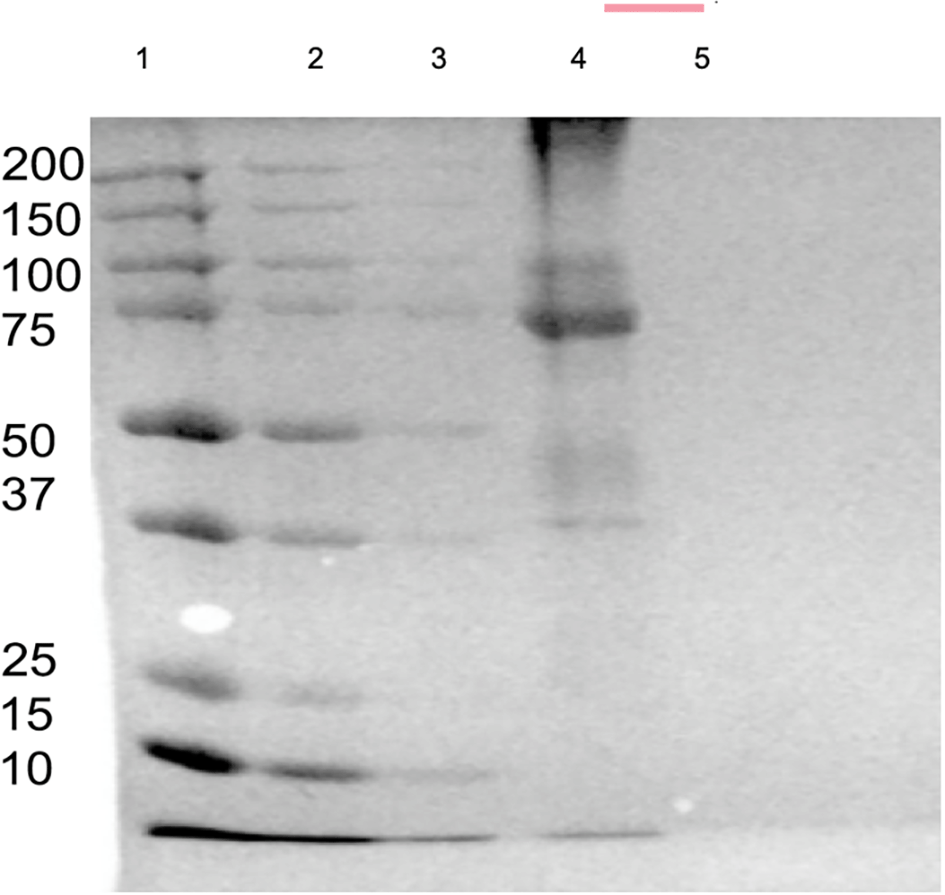
1: The precision Marker, 2.MMLV-RT His-Flow through; 3: MMLV-RT Soluble Fraction; 4: MMLV-RT His Elute; 5. LDS buffer x1.
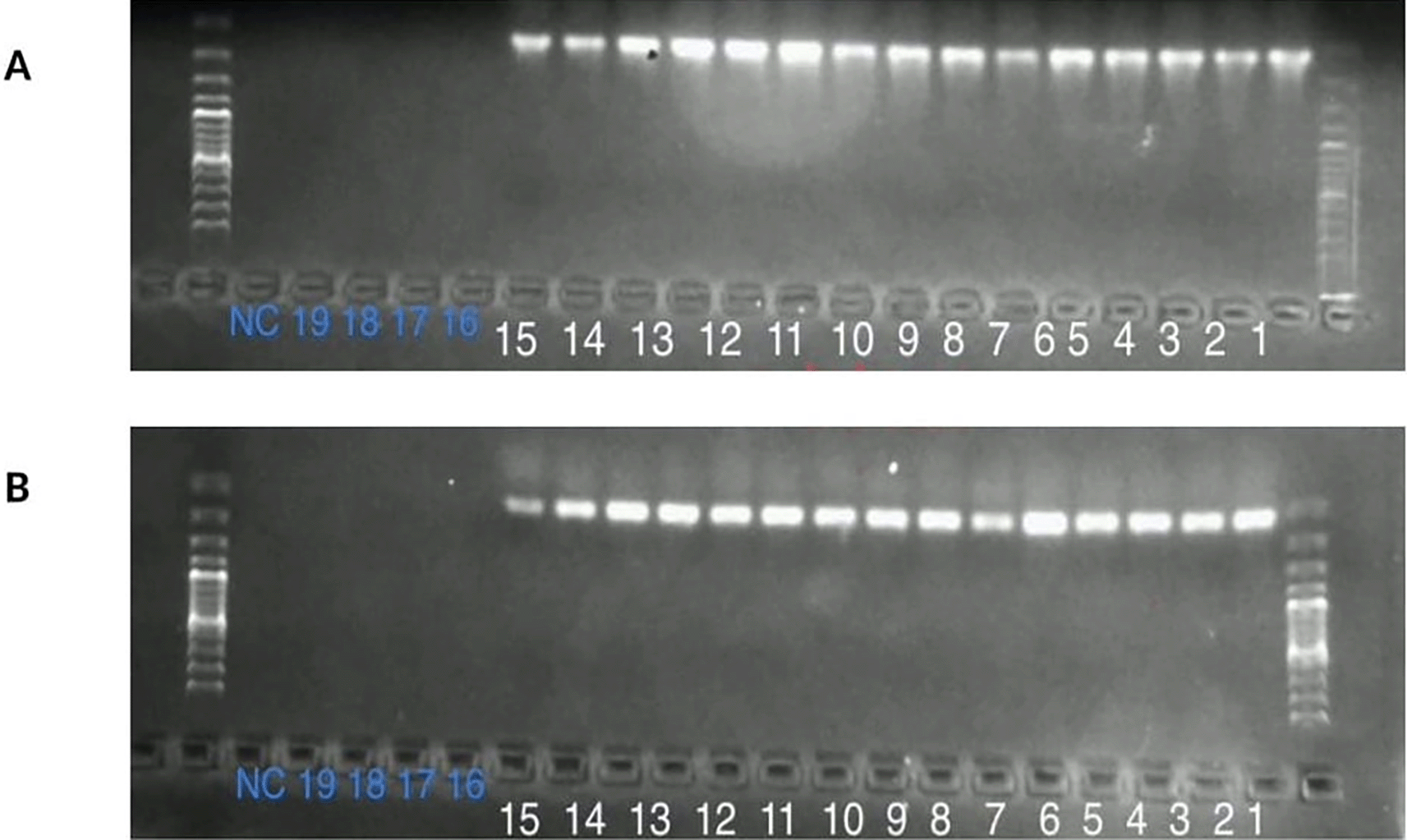
Before testing the performance of the in-house MMLV-RT enzyme, the status of the SARS-CoV 2 samples was verified using real-time RT-PCR, where 89 of 129 were positive and 40 tested negative. Performance testing was performed using commercial Superscript III RT as a control. This was performed using conventional PCR, which included 15 SARS CoV 2 confirmed samples, 4 negative samples, and a negative control. Both enzymes correctly identified 15 positive SARS-CoV 2 as positive. No amplification was observed in negative and negative control samples. The resulting gel is shown in Figure 3.24
To evaluate the developed recombinant In-house MMLV-RT Enzyme, 89 confirmed samples of SARS CoV-2 along with 40 SARS-CoV 2 negative samples and a negative control, were used. RNA extraction was conducted and cDNA synthesis was performed using In-house MMLV RT and conventional PCR. The analysis revealed that the concentrated in-house MMLV-RT enzyme showed a sensitivity of 98.9% (95% CI 0.92-0.996), specificity of 98% (95% CI 0.8712-0.9987). The x10−1 dilution of the enzyme showed a sensitivity of 92.1% and specificity of 95%. The results obtained were compared to those of rtRT-PCR to compute the sensitivity, specificity, PPV, NPV, agreement (Cohen’s kappa), and overall accuracy, as shown in Table 5.24 The receiver operating characteristics were employed to generate the area under the curve, illustrating the diagnostic capability of In-house RT indicated by the sensitivity against the 1-sensitivity curve, as shown in Figure 4.24
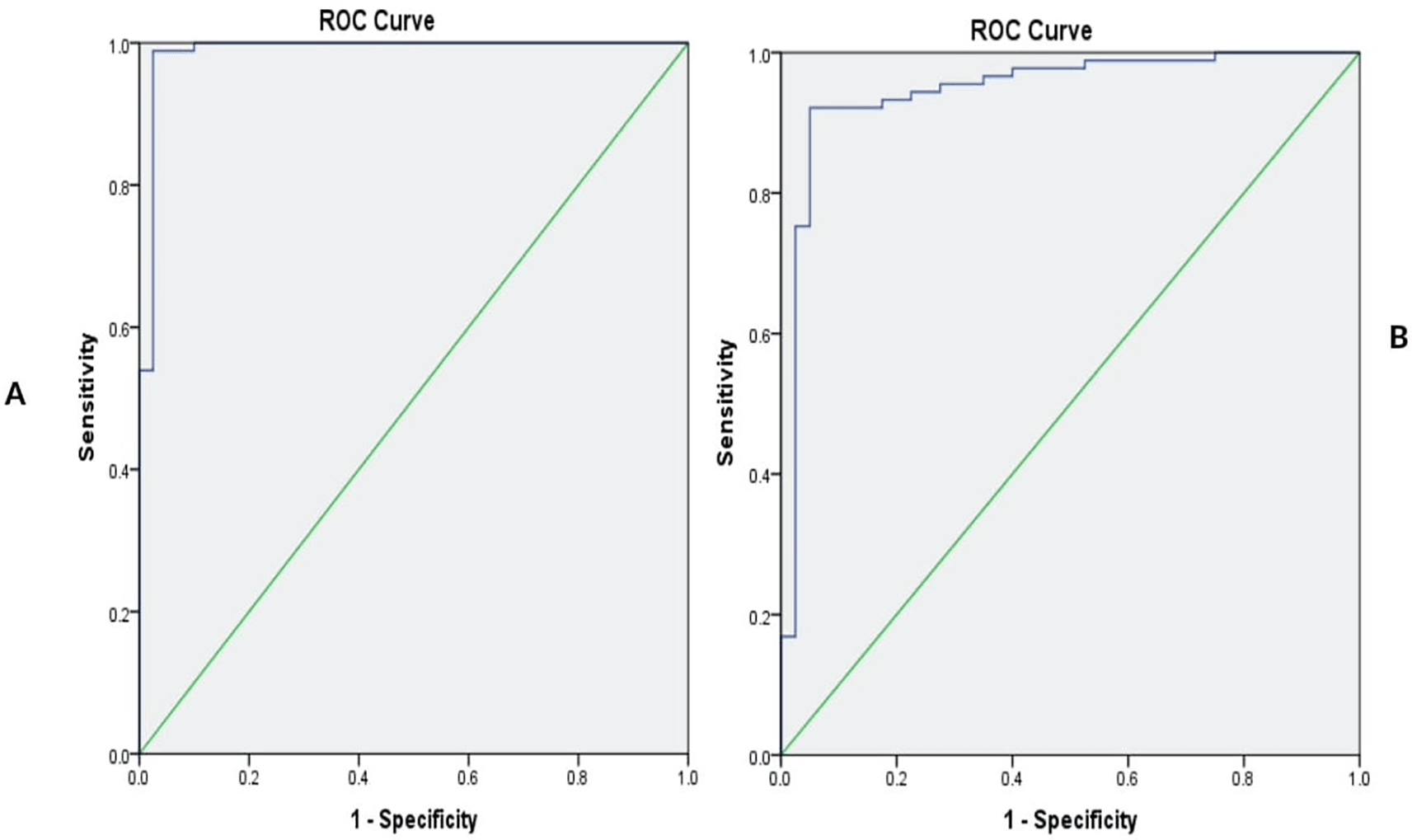
The sensitivity levels of the enzymes are at 0.989(98.9%) and 0.92(92%), respectively.
Reverse transcription PCR (RT-PCR) is a highly sensitive technique that can detect and quantify RNA from a single cell. Unlike Northern blotting and nuclease protection assays, which are commonly used to quantify mRNA levels, RT-PCR requires less amount of RNA. The molecular characteristics of currently used RTs largely affect the efficiency of these reactions.15,16 To optimize their performance, it is essential that these enzymes possess desirable qualities, including high catalytic efficiency at elevated temperatures, ability to synthesize long cDNA products, and a high level of sensitivity.4
This study successfully retrieved and analyzed sequences encoding thermostable Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV-RT). The generation of a consensus sequence enabled codon optimization, which aimed to increase the expression level by substituting the coding sequence of MMLV-RT with synonymous codons encoding the same amino acids for E. coli, enhancing the expression level of MMLV-RT. As reported by Gaspar,17 codon optimization is critical for increasing protein expression and recombinant gene expression in different host organisms. Codon optimization was followed by codon analysis using GenScript Codon Analysis (GenRCA), and the codon Adaptation Index (CAI) was found to be 0.72. The CAI is believed to reflect the expression levels of our gene of interest, and thus, we inferred that the His-MMLV-RT gene would be overexpressed in E. coli given its CAI.18,19 Appropriate GC content can vary depending on the biological context. In certain organisms and genomic regions, high GC content can be advantageous for gene expression and stability; however, in other contexts, low GC content may present challenges related to mutation rates and base quality.20 The average GC content was 57%, which is similar to a previous study.21
Owing to the ease of growth in an inexpensive medium, E. coli is the most popular system for producing recombinant proteins.21 The genetic characteristics of the ECOS-BL21 (DE3) strain, including the absence of proteolytic activity and the presence of a T7 RNA polymerase gene, enable the cloned DNA to be transcribed in high quantities as the transcription rate is high.15 Efforts were made to optimize the culture conditions for protein expression. However, formation of inclusion bodies was observed, which may have been triggered by a high expression rate, as reported by Nuryana.21 In the current study, the protein was successfully expressed after the addition of the IPTG, which increased the expression induction from 3.98 cells/mL to 6.98 cells/mL. These results were similar to those obtained by Nuryana21 and Chen,7 which indicated that the addition of IPTG induced the expression of the desired enzyme.
In comparison to other studies, this study, using SDS-PAGE and Western blotting, demonstrated the quality and purity of the MMLV-RT enzyme.22 The concentration of the in-house MMLV-RT-expressed enzyme was 0.367 mg/mL, as shown in Table 5. After overexpression of the target MMLV-RT gene, the enzyme was first tested to ascertain whether it actually works. This was done by testing the enzyme with 15 SARS CoV 2 positive samples, five negative samples, and a negative control, in comparison with a commercial RT kit, namely, superscript (Invitrogen). Both the commercial and the recombinant in-house RT enzymes showed 15 positive bands and no bands for the five negative samples and one negative control, revealing that the developed enzyme was working.
In the analysis of the performance of the recombinant In-house MMLV-RT Enzyme out of 129 samples analyzed for SARS-CoV 2, 89 positive and 40 negative samples, the concentrated In-house MMLV-RT enzyme showed 87 positive samples, translating to a sensitivity of 98.8%, while the x10−1 diluted In-house MMLV-RT enzyme showed 82 positives, translating to a sensitivity of 92.1%. On the other hand, out of the 40 SARS-CoV 2 negative samples analyzed, the concentrated In-house MMLV-RT enzyme showed 39 negatives, translating to a specificity of 98%, while the dilute In-house MMLV-RT enzyme showed 38 negatives, translating to a specificity of 95%. These results underscore the promising performance of the concentrated In-house MMLV-RT enzyme, affirming its potential utility in SARS-CoV-2 detection with a general sensitivity of 98.9% (95% CI 0.92-0.996) and specificity of 98% (95% CI 0.8712 – 0.9987).
The above results showed that the MMLV-RT enzyme prototype worked in the detection of SARS-CoV 2 RNA as indicated by a p-value of < 0.0001.
Our study has two limitations. First, due to the limited costs, we could not perform field evaluation of the recombinant in-house MMLV-RT enzyme. Second, inclusion bodies were formed during protein expression, which were solubilized during purification.
We successfully produced a recombinant Moloney Murine Leukemia Virus Reverse Transcriptase enzyme whose performance was comparable to that of the standard commercial Reverse Transcriptase, as signified by the P value of the validation process (P value <0.0001), a sensitivity of 98.9% (95% CI 0.92-0.996), and specificity of 98% (95% CI 0.8712 – 0.9987).
AAG: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Project Administration, Resources, Validation, Visualization, Writing – Original Draft Preparation, Writing – Review & Editing; JWK: Conceptualization, Data Curation, Formal Analysis, Methodology, Supervision, Writing – Original Draft Preparation, Writing – Review & Editing; SMN: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Resources, Supervision, Writing – Original Draft Preparation, Writing – Review & Editing; AWM: Investigation, Methodology, Validation, Visualization; FN: Investigation, Methodology, Writing – Original Draft Preparation; GHS: Investigation, Methodology, Validation; JHK: Conceptualization, Data Curation, Formal Analysis, Funding Acquisition, Investigation, Methodology, Project Administration, Resources, Supervision, Writing – Original Draft Preparation, Writing – Review & Editing.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)