Keywords
Schizophrenia, Families, Hierarchical pattern, Common variants, rare variants, Heritability, Biological pathway, Variants
This article is included in the Cell & Molecular Biology gateway.
Schizophrenia, Families, Hierarchical pattern, Common variants, rare variants, Heritability, Biological pathway, Variants
The complicated and devastating phenotype of schizophrenia, coupled with its complex pattern of inheritance, reflects a multigenic pattern and considerable environmental interactions. These interactions are influenced by factors such as psychosocial stress, Toxoplasmosis, cannabis use, and a history of migration and urbanization. Various measures have been used to assess genetic factors, but evaluating the impact of environmental influences has been challenging. Studying the epigenetic background of this disease may be the only valid indicator for understanding these environmental impacts.1
Schizophrenia can be either familial or sporadic, involving many genetic loci that contribute to its genetic heterogeneity. Known popular genes number around 10 to 20 or more, but this varies by population. Multiple loci have been identified in genome-wide association studies (GWAS) and transcriptome-wide association studies (TWAS). The validity of these loci has enhanced the development of polygenic risk scores (PRS). Recent studies confirm the association of PRS with schizophrenia, with an odds ratio (OR) of 1.55 (95% confidence interval (CI) = 1.4–1.7).2
Studying schizophrenia is a good example of examining a complex inheritance disease. In the Sudanese and East African populations, where population diversity is at its zenith and where there is a large effective population size, additional challenges may arise. However, this diversity can also provide a better overview of the genetic components of the disease.3
Recent studies suggest that rare variants play an important role in schizophrenia, offering insights into the disease’s pathogenicity. In a large schizophrenia cohort, whole exome sequencing identified rare variants in 10 genes: SETD1A, CACNA1G, TRIO, SP4, GRIN2A, GRIA3, RB1CC1, HERC1, XPO7, and CUL1. These genes are noted for their high expression in central nervous system neurons and their involvement in synaptic structure and function.4
The overlap between mental conditions poses a significant challenge in psychiatric diagnosis. For example, genetic correlations indicate a high correlation between schizophrenia and bipolar disorder (0.68 ± 0.04), moderate correlation between schizophrenia and major depressive disorder (0.43 ± 0.06), and low correlation between schizophrenia and autism (0.16 ± 0.06).5
The significant heritability of schizophrenia has been a focal point in numerous studies. Researchers have investigated this by examining common variants identified through genome-wide association studies (GWAS), rare copy number variants, and rare variants from exome sequencing studies. Collectively, these genetic factors may account for 30-40% of the heritability of schizophrenia. This underscores the necessity for whole genome sequencing and long-read sequencing technologies to elucidate the complex and heterogeneous genetic background of this disease.6
The evolutionary hypothesis of schizophrenia has been studied in various ways, shedding light on some environmental factors associated with the disease. Recently, prominent evolutionary drives have linked schizophrenia to modern lifestyle factors, including living in developed countries rather than developing ones, and urbanization in general, which is considered a source of chronic stress. Another effective explanation is the association with chronic inflammatory conditions, which could contribute to the etiological understanding of the prevalence of schizophrenia.7
In studying the genetics of schizophrenia, we frequently encounter the question: do common variants contribute to common diseases, or do rare variants predominantly explain the complexity of theses disorders? While our research addresses some rare variants, it emphasizes the necessity of developing new approaches to understand the interactions among multiple variants. Rather than continuously identifying additional variants, we aim to enhance our understanding of how these variants interact, acknowledging that findings often exhibit population-specific, familial, and individual-specific patterns.
This is a case pseudo-control, population-based study encompassing the genomic data of seven families diagnosed with schizophrenia by specialized psychiatrists based on DSM-IV and ICD-10 criteria, each with more than one affected member. Patients were selected for genetic analysis based on being siblings from the same family, with no age or gender specifications. Samples were collected from various psychiatric clinics and hospitals in Khartoum, Sudan, during the period from 2019 to 2021. The total number of participants is 29, including 18 cases and 11 controls.
Whole Exome Sequencing (WES) was performed for one family, while Whole Genome Sequencing (WGS) was conducted for the other six families. The sequencing was done by BGI Tech Solutions (Hong Kong) Co., Limited. The sequencing service included generating at least 90 GB of data per sample, followed by data cleaning.
Basic Bioinformatics analysis was done by BGI analysis team encompassed several steps: data filtering, alignment of reads to the human reference genome (UCSC build HG19), then SNP and InDel calling and annotation of variants to corresponding gene functional units. Data received in Variant Call Format (VCF) files including information about Genes, Transcripts, Function, Variants type, Frequencies and others.
From the Variant Call Format (VCF) files of different variant types, including SNPs and INDELs, filtration of variants shared between all samples from all families, including patients and sibling controls, was performed using Linux. A family-based analysis was then conducted to compare variants shared between patients from the same family but not found in control siblings, using the R program. This comparison was based on variant transcripts or positions and was applied to all variants.
These shared variants, whether between all samples or only between patients, were further filtered according to their impact effect. The impact effects were classified as high, moderate, modifier, and low, based mainly on the variants’ gene functions, excluding intronic, intergenic, or pseudogene regions. Only regions and genes with high and moderate impact effects were included in the final analysis. SNPs and INDELs annotated and send from BGI Company using multiple annotation software for VCF files including clinvar, 1000G, cosmic and other databases.
Five databases were used to analyze the function, clinical impact, and population allele frequencies of mutations: The National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/), Ensembl genome browser 108 (https://www.ensembl.org/index.html), the Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org/), Varsome (https://varsome.com/), and ACMG classification by The American College of Medical Genetics and Genomics (https://wintervar.wglab.org/).
Most of the genes filtered from the previous steps were checked for disease association. Genes already known to be associated with schizophrenia were identified using ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), GeneCards (https://www.genecards.org/), the disease gene database DisGeNET (https://www.disgenet.org/), which contains data about human diseases from literature and GWAS catalogues, and Enrichr - Ma’ayan Laboratory, Computational Systems Biology (https://maayanlab.cloud/Enrichr/).
Each gene was then assessed for related biological pathways to identify any functions linking them to the pathogenicity of the disease using the Reactome database (https://reactome.org/).
The final set of genes was used to draw networks, including genes and protein interactions, using Cytoscape (https://cytoscape.org/), which relies on multiple network databases including GeneMANIA, Uniprot, Reactome, and BioGRID, as well as another network program, ConsensusPathDB (http://cpdb.molgen.mpg.de/).
Tissue expression for single or multiple genes was analyzed using GTExPortal (https://gtexportal.org/home/), providing details about tissue expression of these genes and detecting bulk tissue expression for specific genes accurately by sorting tissues based on median TPM (Median Transcripts Per Million) and displaying gene expression in a heat map view. The analysis is summarized in a flowchart (Figure 1).

Form these families patients already diagnosed and on treatments, the main age group is (20-40) years old, there are no significant gender differences 10 females and 8 males.
Family 1 (F1): Both patients are males undergoing treatment. Their age at diagnosis was 17 and 18 years old. Their elder brother, who is healthy and free from mental disorders, served as the control. Additionally, two sisters from the same family have learning disabilities, with one showing typical hallucination and delusion symptoms but remained undiagnosed due to financial constraints. Both patients in F1 had poorly controlled symptoms despite medication.
Family 2 (F2): This family includes four affected members: two sisters, one brother, and their mother. Sequencing was successful for two affected sisters and two controls: their elder brother and younger sister. The two affected sisters’ age at diagnosis was 10 and 25 years old.
Family 3 (F3): Two affected members—a male and a female—and their healthy elder brother were sequenced.Their age at diagnosis was 35 and 25 years old, respectively. Both patients had poorly controlled symptoms despite antipsychotic medication.
Family 4 (F4): This family has two affected male patients. Their age at diagnosis was 20 and 24 years old, the elder patient also suffers from diabetes with associated complications, while the younger patient showed good compliance with medications. They have a control brother.
Family 5 (F5): Four affected siblings—three males and one female—and one unaffected sister as control. Their age at diagnosis was 17, 21, 23 and 27 years old. The father occasionally experiences delusions and paranoid ideas but has never been diagnosed with schizophrenia.
Family 6 (F6): Four affected female patients and their elder brother as control. Their age at diagnosis was 18, 33, 35 and 36 years old. The eldest sister showed severe symptoms with a long duration of illness and poor medication control, whereas the other patients had shorter duration and milder symptoms upon diagnosis.
Family 7 (F7): This family consists of two affected sisters as patients and their elder brother as control. They are the youngest siblings in the family, and both sisters have shown a good response to medications. Their age at diagnosis was 21, and 23 years old.
Results were categorized and analyzed hierarchically based on their frequency within the families and their potential contribution to the phenotype (Figure 2). The flowchart summarizes data ranging from common variants shared between patients and family controls to rare variants.
The presentation of results depends on the degree of sharing between cases, with a focus on variants classified as having high impact effects. These variants are further categorized based on allele frequency, whether they are either common, or rare variants, with minor allele frequency (MAF) less than 0.01 or more than 0.05 (0.01< MAF <0.05) to assess their potential contribution to the phenotype.
Initially, all 29 samples, including both schizophrenic patients and sibling controls, were analyzed together. Assuming that the control sibling may carry the mutant allele without manifesting the condition. From the variants shared across all samples, 47 high impact SNPs and 42 high-moderate impact INDELs were selected. Most of these prioritized common variants are benign with some considered as likely pathogenic or of uncertain significance according to ACMG classification. Some already known to be associated with schizophrenia.
The analysis of shared SNPs among all samples, including both patients and sibling controls, identified 47 variants with high impact mutations. These mutations are associated with functions such as structural interaction variants, stop lost, splice donor, splice acceptor, and stop gained, suggesting their relevance to schizophrenia within these families.
Most of these variants exhibit a high frequency of the ancestral allele (1-0.8), with two exceptions noted in SND1 and NCAM1, where their frequencies are 0.6. NCAM1 is known to be associated with schizophrenia and is overexpressed in the frontal cortex of the brain.
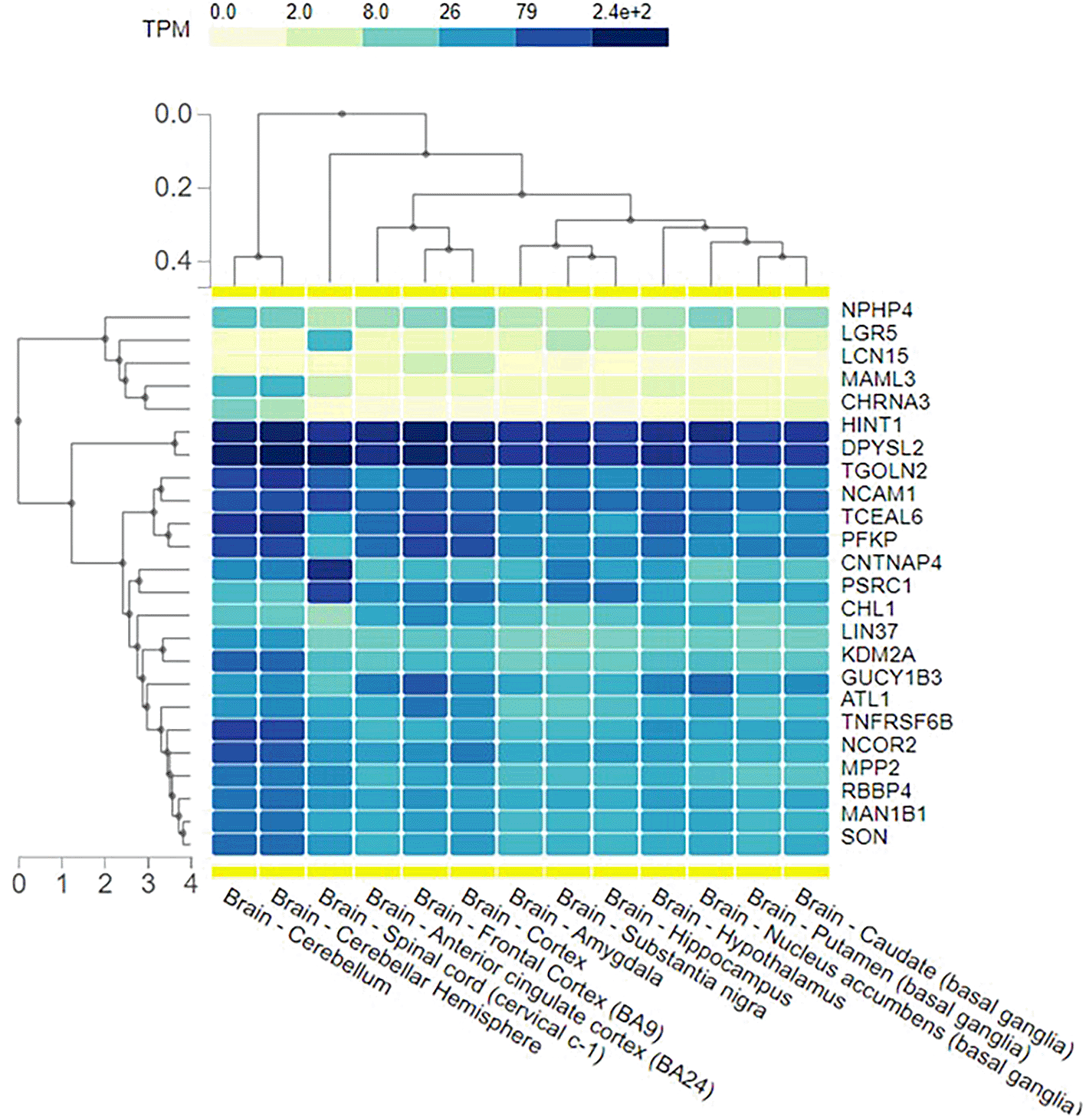
The majority of these genes show expression in the brain, primarily in the cerebellum, frontal cortex, and spinal cord (Figure 3).
Symbols/Abbreviation: SNPs: Single nucleotide polymorphisms;
INDELs: Insertion/Deletion.
25 variants from the common variants group were identified despite being shared in all cases and the small sample size, their selection based on their high impact classification according to our sequencing annotation and their brain expression.
Most of these variants are found in genes show high brain expression, the majority expressed in the brain frontal cortex, while another eight genes are highly expressed in the cerebellum (Table 1).
Among these genes, DPYSL2, MPP2, ALDH3B1, NCAM1, CHRNA3 and GPRIN2 are known to be associated with schizophrenia, whereas NCOR2 is associated with bipolar disorder according to disease gene databases.
Comparison of the shared variant frequencies between the general population and Sudanese population showed almost no differences confirming that these variants are common in both populations. However, there are exceptions among four INDEL variants (rs11284059, rs11448549, rs34377180, and rs34373121), which show significant lower frequencies in the Sudanese genome (0.38-0.44) compared to the general population.
Family based analysis including rare variants: Variants collected from all families included 388 SNPs and 188 INDELs, and we selected 41 genes containing both SNP and INDEL variants. The selection was based on variants being shared between patients and absent in controls within each family, and then being shared between at least two families, regardless of having the same variant ID. Importantly, there were no variants shared between all seven families. There was a significant difference in the presence of high impact variants between cases and sibling controls in families, with a P value of 0.00862. From this group, we focused only on high-impact rare variants and further narrowed down to ultra-rare variants with an allele frequency ≤ 0.05%.
One INDEL variants shared between all samples including cases and controls presented in Table 1, irrespective of allele frequency, in NCOR2 (rs143952466) classified as rare variants with allele frequency assessed by gnomAD database (AF = 0.00002060) mainly expressed in Pituitary gland and Cerebellum. Associated with Bipolar Disorder according to diseases gene database. Classified by ACMG as uncertain significance.
Most of these rare variants are present in two or three families and are classified as likely benign/benign according to the ACMG classification. However, 12 variants were considered of uncertain significance and are located in genes associated with schizophrenia according to the OMIM and ClinVar 2019 databases. This group of genes is associated with Neurotransmitter Clearance according to the Reactome database, with a P value of 0.03475, suggesting a potential link to schizophrenia. However, the most highly involved biological pathway is the Biosynthesis of Maresins, which is more likely associated with type 2 diabetes.8
Other genes shared between families with more than two variants regardless of being rare variants, including both SNPs and INDELs, are CYP2C8, MUC6, PABPC3, COMT, and PADI4. Most of these variants are classified as benign by ACMG classification.
Genes known to be associated with schizophrenia include the COMT gene, which presents a shared mutation in four variants, one of which (rs199710929) is ultra-rare with an allele frequency of 0.0001402. The PADI4 gene has three variants (rs874881, rs2240335, rs2240335) with an allele frequency of ≤ 0.5%, and the CTNNA2 gene shows the same variants in two families (rs7609047, AF = 0.2435). The NAALAD2 SNP (rs139259253) has a rare allele frequency of 0.00008843 and is classified by ACMG as of uncertain significance. Additionally, the SNTG1 gene (rs770648099, AF = 0.2098) is highly expressed in multiple brain regions, with the highest expression in the brain’s frontal cortex.
The MUC19 gene presented with five different variants, including two SNPs, two INDELs, and one deletion (SVs). The most interesting variant is a rare SNP (rs73100424) with an allele frequency of 0.0415, which is functionally classified as of uncertain significance.
The total rare variants, some of which were mentioned previously, include ultra-rare variants with an allele frequency of ≤0.1%. None of these variants are pathogenic, but 12 are considered of uncertain significance according to ACMG classification. These variants are located in the following genes: ST6GALNAC1, COMT, CYP2D6, NAALAD2, CDNF, PRSS3, PRAMEF2, ZDHHC11, TAS2R46, CYP2C8, NBPF9 and OR5K4 (Supplementary Table).
The gene network includes shared genes between all samples with high impact common variants revealed 14 important central genes. Among these, SON and NCAM1 have been mentioned previously due to either their lower frequency or their likely pathogenic status. The main centrality genes include RIPK1, RBBP4, MAP 3K1, NCOR2, and ZNF2. Only SON, CHRNA3, and DPYSL2 are known to be associated with schizophrenia. Other genes in the network include TAAR9, CREB3L1, NET1, LTF, and VEGFC. Most of these variants are expressed in the brain, but unlike the known cerebral regions associated with schizophrenia, there is high expression in the cerebellum (Figure 4).
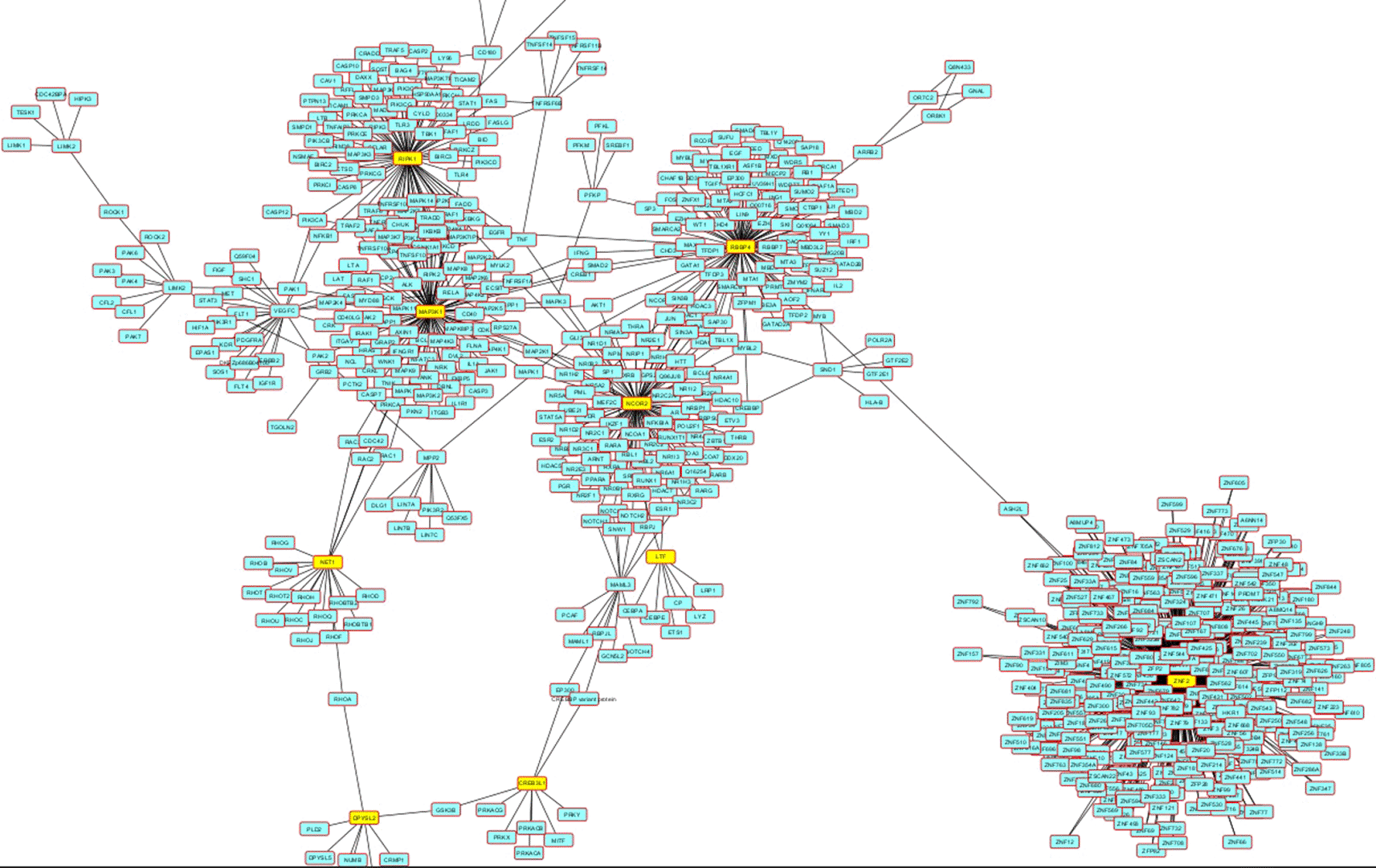
By using the STRING database and creating a more extensive network involving the previously mentioned genes, the expanded analysis revealed the involvement of additional genes. Together, these genes are related to three schizophrenia-associated biological pathways: catecholamine biosynthesis, metabolism of serotonin, and enzymatic degradation of dopamine by monoamine oxidase.
Other genes were studied because they are located at the center of each family network, and also appear in the network done by cystoscope joining all rare family variants. Genes were selected based on being shared between patients but not family controls. Three of these centrality genes are known to be associated with schizophrenia: PTPN6, which is central in two family networks with similar variants (rs2110072, AF = 0.9477) related to the immune system and cell communication; CTNNA2 (rs7609047, AF = 0.2534), associated with apoptotic cleavage of cell adhesion proteins, potentially contributing to neurodegeneration and neurodevelopmental issues; and GRIA2 (rs34460606, AF = 0.004842), which is central in the gene network and can contribute to the pathogenicity of schizophrenia due to its involvement in the activation of NMDA receptors, postsynaptic events, and neurotransmitter signaling pathways. Other genes from different family networks known to be associated with schizophrenia include HLA-B, HDAC9, PTPN1, TBP, and DGKH. Additionally, MAP 2K3 (rs55796947) and TMEM216 (rs10897158) both presented with SNPs that are not rare but are classified as likely pathogenic as germline mutation by Varsome data base.
In each family shared genes networks also in all families rare variants network, there is a cluster of ZNF genes interacting within a large group of the same gene family, but not interacting with other gene families. For the general rare variants network the ZNF717 was the centrality gene which is not known to be associated with schizophrenia.
Schizophrenia is a highly heritable condition with a suggested 1% prevalence in all populations.9 Multiple evolutionary theories have been presented to explain the persistence and selection of schizophrenia genes despite the low reproductive fitness of affected individuals. Some view this paradox as a tradeoff in human evolution linked to brain development after divergence from earlier ancestors like the Neanderthals, associated with the evolution of language, social skills, and cognition. Studies in this field suggest that schizophrenia-associated genetic loci have undergone recent positive selection in humans after the divergence of modern humans from Neanderthals.10
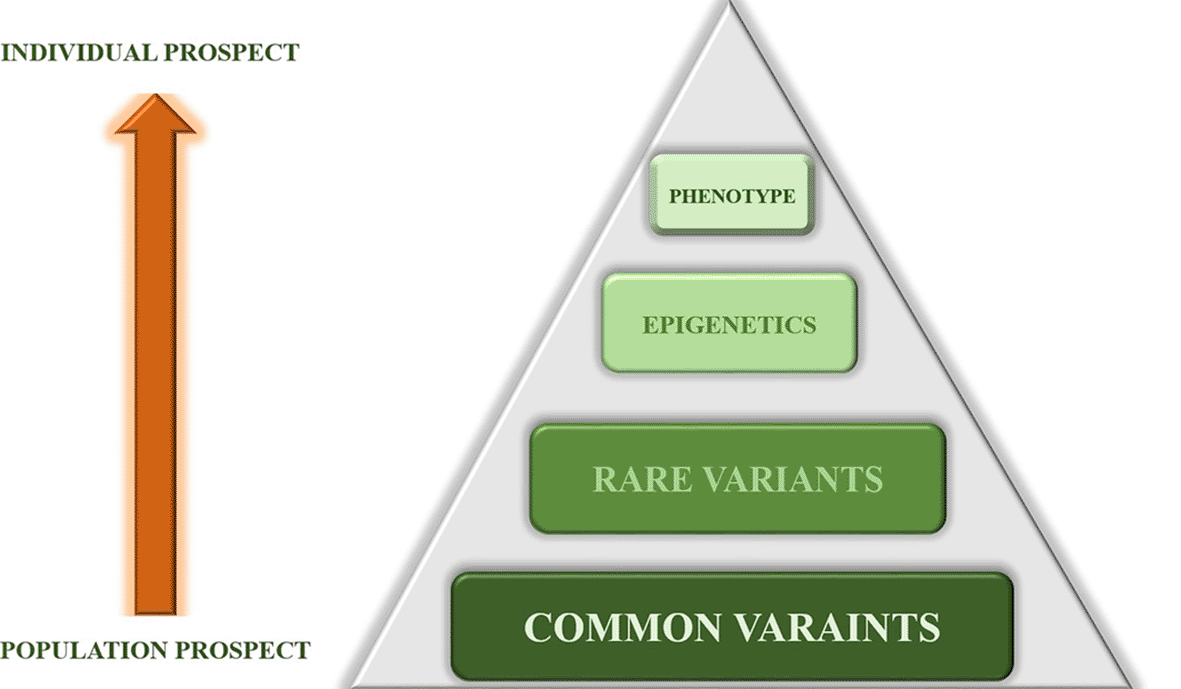
In the current study, we suggest a novel analytical approach where all variants in the population and individual are combined in a hierarchical manner to provide a comprehensive explanation of the phenotype.
Variants were categorized into common and rare groups representing the first and the second steps of the hierarchy. We studied each group separately, starting with the common variants. We began by examining 47 high-impact variants conserved in schizophrenia families, which show the highest frequency of ancestral alleles in the African population in general, in tandem with the recent African origin of modern humans. This approach involves including both cases and sibling controls variants, based on the assumption that the controls may harbor some of the pathogenic variants without manifesting the phenotype.
Most of the identified genes were expressed in the cerebellum. Although the cerebellum’s involvement in schizophrenia has been studied, and many approaches suggest its role in disease pathogenicity, the direct functional changes may be minimal. Nonetheless, the cerebellum is part of neuronal circuits related to multiple cortical functions and higher cognitive functions. An important approach proposes that the neurodevelopment of the cerebellum is relatively late and may coincide with the onset of schizophrenia, which supports the neurodevelopmental theory of the disease.11
This group of genes with common variants collectively suggests multiple significant biological pathways associated with schizophrenia according to the Reactome database, including the L1CAM Interactions pathway, Axon Guidance-related pathways, and Nervous System Development. These findings strongly support the hypothesis that schizophrenia arises due to neurodevelopmental dysfunctions in the brain, potentially involving abnormalities in neuronal cell adhesion molecules, axon guidance, synaptogenesis, and cell migration. All these events contribute to neuronal circuit malformation and the development of schizophrenia.12,13
Most of the interesting genes in our data, in one way or another, support the neurodevelopmental theory of schizophrenia and link schizophrenia-associated genes to multiple brain processes specific to the modern human brain, reflecting its evolution and function.
The premise that schizophrenia is a neurodevelopmental disease can also be explored at the next level of the hierarchy by analyzing each family separately, focusing on variants with low frequencies, and considering the specific genetic makeup of each family beyond their similar clinical backgrounds and manifestations. Multiple genetic studies aim to unravel the complexities of psychiatric conditions, particularly schizophrenia, by identifying risk alleles that vary between populations and individuals. Rare variants detected in such complex disorders are considered useful tools for identifying pathogenic genes and studying individual risk alleles as part of personalized medicine.4
However, no rare variants were shared between all cases and no controls in families indicate that the complex etiology of schizophrenia might be unique to each family and indeed of individualized nature. The gene NCOR2 was found to be shared among all samples, regardless of their classification as cases or controls. This could be explained by assuming that being a sibling to a patient with a complex inherited disease may expose one to disease-associated variants. The gene exhibits an insertion-deletion (INDEL) variant (rs143952466) with an ultra-rare allele frequency (AF = 0.00002060) and is expressed in the brain. According to the Reactome database, NCOR2 is involved in the loss of methylated CpG binding protein 2 (MECP2) binding ability to the NCoR/SMRT complex, which regulates its expression and activity. Recent studies suggest that NCOR2, through its interaction with MECP2, may be associated with neurodevelopmental disorders, including autism.14
An overview of the schizophrenia-associated genes from our seven Sudanese families reveals that there is no simple scenario for the disease, and that genetic complexity prevails both within and between families from the same population. No single variant was shared across all cases without also being present in controls. However, genes with multiple variants could be shared among two or more families. Another observation is that common variants known to be associated with schizophrenia are not predominantly represented in this study, with a few exceptions, such as the COMT gene. Additionally, rare variants identified in this study differ from those found in other populations, showing better association with data from African descendants compared to European descent4.15 This finding is not entirely surprising given the complex genetic underpinnings of the disease and the diverse genetic makeup of the Sudanese population, where population structure plays a significant role in this complexity. From this hierarchical analysis, which includes most of the genome data, we can postulate that all these variants collectively contribute to the genetic background of susceptibility to this disease.
The rare variants are generally associated with multiple biological pathways, including interferon-gamma signaling and neurotransmitter clearance. MECP2 regulates neuronal receptors and channels. According to ClinVar, these variants are primarily associated with schizophrenia, as well as Parkinson’s disease and type 2 diabetes mellitus.
From both common and rare variant types, gene network analysis suggested the involvement of multiple centrality genes, including variants associated with schizophrenia. A common Biological pathway prediction using Reactome database shed light on prevalent biological pathways, prominently featuring “Defective RIPK1-mediated Regulated Necrosis.” This pathway, associated with necroptosis, a vital programmed cell death mechanism alongside apoptosis, encompasses diseases like neurodegeneration and autoimmune disorders.
Reviewing literature, the apoptotic subtype in the central nervous system is identified as necroptosis, mediated by genes such as RIPK1, RIPK3, and MLKL. This process triggers neurodegenerative events observed in conditions like Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and amyotrophic lateral sclerosis.16
In conclusion using these variants together may referred to the classical neurotransmitter theory of schizophrenia, highlighting changes in dopamine, serotonin, and other receptors. Another significant clue points to immune-inflammatory effects, which support neurodevelopmental and neurodegenerative explanations for the disease.
At this level, we face important questions about how gene expression is affected and how environmental factors—such as stress, social disabilities, lifestyle, and others—contribute to the phenotype. Understanding these factors, along with epigenetic determinants, is a crucial third step in this hierarchical analysis, which unfortunately is missing from this research. Epigenetic determinants, including DNA methylation, histone modification, and non-coding microRNAs, are essential for answering major questions about their role in psychiatric conditions. For example, stress is a known factor in PTSD, and in schizophrenia, a broad spectrum of environmental factors, such as infections and cannabis use, can be relevant.17 Additionally, epigenetics is important for understanding the overlap between psychiatric conditions, both genetically and phenotypically, which may ultimately lead to the reclassification of these diseases based on not only clinical presentation but also genetic background. By exploring the progression from various types of genetic variants to epigenetic factors, we can gain a more complete picture of the phenotype, making diagnostic and therapeutic approaches more individualized and effective.
To conclude, there is an ongoing debate between the common and rare variant hypotheses in understanding the genetic basis of complex inheritance diseases. The common variant hypothesis posits that a few common genetic variants can significantly contribute to disease pathogenicity, while the rare variant hypothesis suggests that multiple rare, low-frequency variants may have a stronger impact.18 It is important to note that both exhibit limitations in fully explaining the heritability.
It is time to consider both approaches within a hierarchical model that integrates these variants along with epigenetic factors and their interactions. This system-based approach shifts the focus from merely counting variants or assessing their frequencies to understanding their broader, interactive effects on disease development. This approach faces significant limitations, including a small sample size, the lack of an epigenetic profile, and the absence of a statistical model to confirm the proposed hierarchical pattern.
The project has been approved by the National Research Ethics Review Committee of the Health Research Council, Federal Ministry of Health, Republic of Sudan in 22/10/2019 under proposal no. (5-10-19) entitles (Whole genome sequencing in analysis of schizophrenia patients and their antipsychotic drug response variation in Sudan) submitted by Dr. Aza Elshafie Khidir Saeed, of the Institute of Endemic Diseases/University of Khartoum.
Patients were sampled and interviewed by the principal investigator following psychiatrist approval. Written informed consent was obtained from the patients themselves or their guardians after providing an informative description and discussion and approved by National Research Ethics review Committee of the Health Research Council, Federal Ministry of Health, Republic of Sudan.
Azza Saeed
Role: Conceptualization, Formal Analysis, Funding Acquisition, Methodology, Validation Writing Original Draft. Review & Editing the manuscript.
Lamees Ahmed, and Modathir Salih
Role: Formal Analysis and software program
Safa Abuswar
Role: Visualization
Amel Eltigani
Role: Investigation
The psychiatrist responsible of clinical diagnosis and patient’s evaluation
Ayman Hussein
Role: Project Administration
Schizophrenia project principle investigator
Magdeldin Elgizouli
Role: Writing, Review & Editing
Muntaser E. Ibrahim
Role: Supervision, Conceptualization, Writing, Review & Editing
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)