Keywords
Glioblastoma, residual disease, patient-derived models, 3D models, replacement.
This article is included in the NC3Rs gateway.
Glioblastoma, residual disease, patient-derived models, 3D models, replacement.
Scientific benefits:
• Unique access to the most brain-invasive GSCs feasible in living patients.
• Patient-matched comparison of biology in GSCs from resected and typically residual regions.
• Avoidance of cell selection based on metabolic features (including 5-ALA metabolism) during derivation.
• Detailed and regularly updated associated clinicopathological data.
• Embeds intra-tumoural heterogeneity assessment early in translational pipelines to ensure only treatments with activity against residual disease are progressed.
3Rs benefits:
• Reduce use of animal-based preclinical models through improved drug candidate triage.
• Replace use of animal-based preclinical models to fast-track candidate therapies within drug repurposing strategies (from both other cancers and other conditions).
• More rapid end assay data generation compared to animal studies.
Practical benefits:
• Compatible with commonly used laboratory assays.
• Robust 3D clonogenic survival assays.
• Over 44 resected and residual GSC models available for academic use via MTA for groups wishing to replace/reduce PDX model usage.
Current applications:
• Focused functional and omic profiling of the differences between GSCs from tumour core and distant invasive edge regions.
• Evaluation of spatially-mediated differences in therapeutic response.
• Preclinical assessment of therapeutic efficacy in normally ‘left-behind’ disease.
Potential applications:
Glioblastoma is a devastating incurable brain cancer that contributes to around 190,000 brain tumour related deaths/year globally.1,2 Typically, surgery is performed to remove as much of the tumour as safely possible. However, the highly infiltrative nature of glioblastoma underlies a lack of distinct tumour-brain interface intraoperatively and ensures residual disease at a macroscopic or microscopic level is inevitable.3 Subsequently, patients are treated with radiotherapy, temozolomide chemotherapy and increasingly tumour treating fields (TTFields).4 Unfortunately, despite current multi-modal therapy, disease progression occurs within 6-7 months on average and less than 10% of patients survive more than 5 years.5,6
The failure of preclinical research over the last decade to portend truly substantial improvements in patient survival for glioblastoma, a recognised cancer of unmet need,7 implies that commonly used preclinical models do not fully reflect the complexities of tumour cell biology and therapeutic resistance. Therefore, efforts to increase the clinical and surgical relevance of early preclinical research models may not only enhance our understanding of difficult to treat cancers, but also improve the predictive value of preclinical studies to delineate therapeutic strategies which might confer meaningful benefit for patients.8 In the context of glioblastoma, inherently treatment resistant glioma stem cell (GSC) subpopulations have been identified to exhibit heightened DNA damage response (DDR) activity following current chemoradiotherapy9–11 and contribute to tumour repopulation and disease progression.9,10,12,13 Furthermore, beyond clear patient-to-patient variation,14–16 extensive spatial heterogeneity within each individual tumour contributes to the therapeutic recalcitrance of glioblastoma.17–19 However, both in vitro and in vivo research models used in the laboratory normally do not represent the spatial complexity of human glioblastoma observed in patients.
Rodent (particularly mouse) orthotopic patient-derived xenograft (PDX) models are commonly used in glioblastoma research20 and are often viewed as a gold-standard model to investigate candidate therapies prior to clinical trials.21,22 Based on our recent analysis of studies using murine PDX models as patient-specific avatars to evaluate treatment response, in conjunction with data obtained from the GlobalData database detailing current therapeutic development for glioblastoma, we estimate over 20,000 mice were used globally in the setting of patient-specific treatment response prediction over the last 5 years.23 More generally, over 630 research articles published in 2023 mention the use of a glioblastoma animal model (PubMed). Reviewing of a sample of these studies, we find approximately 120 animals are used on average per study. This extrapolates to a conservative estimate of over 75,600 animals per year being used in glioblastoma research globally.
Standard practice in orthotopic PDX models includes the placement of tumor cells into the brain parenchyma of immunodeficient mice or rats, typically leading to observable disease progression and ultimately necessitating animal culling. Numerous advantages are provided by these models including, to some degree, recapitulation of the blood-brain barrier, generation of therapeutic response data, and some information around treatment tolerability. However, despite broad use by the research community, these models are inherently flawed. Orthotopic and subcutaneous tumor implantation in PDX models of glioblastoma, when viewed through the lens of the 3Rs, present ethical challenges due to the harsh procedures involved. To evaluate therapeutic efficacy, tumor growth is often permitted to progress to a specified volume before commencing treatment, with many animals still undergoing symptomatic disease progression and ultimately requiring euthanasia. This approach classifies the studies as 'severe,' reflecting the unavoidable distress, discomfort, and non-survival of the rodent subjects. The time it can take to generate meaningful data using PDX models also represents a further disadvantage. The duration between patient tumour resection to generation of in vivo therapeutic response data using PDX models can take well over a year, as time is required to: passage and expand tumour cells either in vitro or in vivo (rodent-to-rodent) to provide a cohort large enough to evaluate one of multiple treatment strategies/conditions, and, reach a predetermined tumour size/volume before starting treatment. Even where a pre-existing cell line is implanted orthotopically, many months can be required for tumours to grow to an acceptable size prior to treatment. Consequently, we believe the development and implementation of ex vivo cell culture models of residual disease that are more representative of the disease left behind after surgery in patients has the potential for broad impact and defined scientific advantages over animal models (discussed below). These models therefore have the potential to reduce animal usage and replace animal studies in a number of contexts whilst mitigating against some of these flaws.
From a scientific perspective, orthotopic PDX models of glioblastoma do not accurately reflect the molecular profile and full spectrum of heterogeneity in patients for numerous reasons. For example, animal-specific evolution of glioblastoma cells following implantation has been reported.24 Furthermore, orthotopic rodent xenografts generated through implantation of either established or more recently patient-derived cell lines often form tumours with negligible-to-limited brain invasion - unlike the highly brain invasive tumours observed in patients.25 To some degree, this may reflect spatial sampling bias since the overwhelming majority of research tissue used to generate these implanted glioblastoma cells will be liberated from the typically resected tumour bulk, rather than more distant, brain invasive disease that is more likely to be left behind after surgery.8
Moreover, due to the technical challenges and infrequency of surgical resection in rodent models, these xenograft systems often fail to replicate the full spectrum of multi-modal therapies routinely administered to glioblastoma patients. As a result, it remains uncertain whether the observed survival benefits in most orthotopic PDX models represent treatment effects on the resectable tumour core, or if they extend to the infiltrative tumour margins that drive disease recurrence in patients. This is clinically relevant, as in patients receiving standard-of-care treatment, only the infiltrative residual cells left behind after surgery are responsible for disease progression and death. In many contexts, such as attempts to apply therapies used for other cancers to glioblastoma or repurposing drugs used to treat other medical conditions, an in vitro alternative with the capability to directly assess treatment response in residual cell populations could provide a more meaningful and clinically-relevant replacement for current animal studies. In the assessment of newly developed therapeutics, in vitro assessment of drug response in residual disease cell populations could help triage candidate drugs – ensuring only those with activity against residual disease progress to in vivo studies to minimise animal usage.
The translation of innovative glioblastoma therapies from bench to bedside has largely hinged on the use of cells obtained either recently (e.g., primary and ex vivo cultures) or long-established (e.g., immortalized cell lines), all originating from the main bulk of patient tumours that is typically resected during surgery. In light of the limited survival improvements in glioblastoma compared to other cancers, the traditional framework—relying on 2D in vitro studies and subsequent xenograft models with resected cells—has proven inadequate in predicting the response of the brain invasive glioblastoma cells that remain after surgical intervention in the clinic. The expanding arsenal of targeted therapies underscores the necessity for models that more faithfully represent the biology and therapeutic response of post-surgical residual glioblastoma early in the translational research process8 to accelerate the application of next generation therapeutic strategies and combinations to future patients diagnosed with glioblastoma. To address this unmet need, we have developed robust, adaptable ex vivo models of post-surgical residual glioblastoma from human tissue which can be readily used within 2-dimensional and 3-dimensional culture conditions, including within a customised scaffold-based architecture.26,27 These models aim to enrich surgically-relevant investigation of the complex biology of glioblastoma within early preclinical studies, with the potential to replace animal studies in some of these contexts.
Surgical resection, when guided by the anatomical specifics of a case, may occasionally result in the extraction of a large en bloc specimen (such as a partial lobectomy), incorporating areas of infiltrated brain that would typically evade resection. We believe that this tissue provides a crucial resource in modelling and understanding residual disease in glioblastoma. Our ex vivo resected and residual models are based on the generation of parallel, patient-derived GSC lines from the tumour core and adjacent infiltrated brain of suitable specimens. Here, we outline the protocol used to successfully derive parallel, spatially-distinct resected and residual glioblastoma models that can be maintained as either bulk (differentiated) or GSC cultures for a wide range of preclinical research applications to study the unique features of post-surgical residual disease. Crucially, these models may provide a biologically relevant and cost-effective alternative to the use of animals in some preclinical research. For example, they provide a particularly compelling replacement for murine models to assess therapeutic response for treatments which have already progressed beyond phase 1 clinical trials for other types of cancer and/or repurposed medications. Therefore a conservative target of 10% uptake of our ex vivo residual models within glioblastoma research community internationally, with a focus on these settings, could replace the use of over 7,000-8,000 animals per year (based on usage leading to PubMed indexed publications, estimated above), whilst accelerating the translation of comprehensive novel therapeutic strategies into the increasingly adaptive outlook of later stage glioblastoma clinical trials, e.g. The Tessa-Jowell BRAIN-MATRIX trial, 5G and GBM AGILE.1,28
The procurement of en bloc specimens suitable for resected and residual model generation requires appropriate institutional research ethics. At our institution, all clinical information and specimen acquisition was performed in within the scope of the approved IRB ethics protocol 11-YH-0319 (STH15598) - Yorkshire & The Humber - Leeds East Research Ethics Committee (approved 03/11/2011, substantial amendment approved 01/06/2016) and in adherence to the Declaration of Helsinki (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/). Close collaboration across academic and clinical boundaries with input from the neuro-oncology multi-disciplinary team (MDT) is essential. In our experience, the availability of clinical-academic surgeons with experience of navigating both environments has been incredibly useful. However, in the absence of this, ensuring some members of the research team have appropriate approvals to access both the laboratory and neurosurgical operating theatre is vital to visually confirm the correct orientation of specimens is maintained during the transfer process from the clinic to the laboratory. Additionally, clear and regular communication with the theatre team including the theatre team leader, scrub nurses, operating department practitioners, anaesthetists and surgeons is key to maximising successful procurement of these relatively rare samples.
1. Case selection
1.1. ln collaboration with an appropriately qualified neurosurgeon, identify suitable patients based on the inclusion and exclusion criteria detailed in Table 1. Typically, potential candidates are those scheduled for surgical resection of a large suspected high-grade glioma situated anteriorly within the frontal or temporal lobe with a planned resective strategy that is likely to produce an en bloc specimen, usually as a partial lobectomy (see Figure 1A). Importantly, sample acquisition is opportunistic with availability dictated by the tumour characteristics and operating neurosurgeon(s); the research team must never attempt to influence the resective strategy planned by the clinical team.
NOTE: We find weekly clinician/academic-clinician review of cases referred into the neuro-oncology MDT (tumour board meeting) and scans of scheduled neuro-oncology cases invaluable to identify cases as early as possible and provide sufficient time to obtain patient consent.
1.2. Confirm lead/operating neurosurgeon responsible for the patient’s care is supportive of a suitable patient being approached for consent.
1.3. Obtain patient consent as per agreed institutional protocol.
2. Macrodissection
NOTE: Prior to the expected time of specimen retrieval, in the laboratory, disinfect and prepare a class II biological safety cabinet ensuring the availability of the required reagents, plasticware and equipment listed in Tables 2-4 and start warming the following solutions to 37°C: PBS, Accutase, complete GSC media (see Table 5).
2.1. Ensure that a member of the research team is present in theatre as the tumour is being resected.
2.2. Confirm freshly resected en bloc surgical specimen is suitable for parallel resected and residual model derivation in close collaboration with the neurosurgical and/or histopathology team (based on institutional set-up/approvals) – ensuring the specimen contains clear macroscopic evidence of tumour and adjacent tissue with cerebral cortex visible.
2.3. Neurosurgeon (or histopathologist based on institutional processes) divides en bloc specimen along its axis to yield a specimen for clinical histopathology and a separate specimen for research both containing tumour core and adjacent brain tissue.
2.4. A member of the research team immediately transports the pseudonymised research specimen to the laboratory in a clear specimen container within clear specimen bags, whilst maintaining orientation of the sample.
Tip: Although we typically find use of transport media/solution unnecessary as our neurosurgical theatres and research laboratory are in close proximity, if desired, research specimens can be transported in sterile normal saline or advanced DMEM/F12 media at room temperature.
NOTE: At our institution, research samples are pseudonymised by assigning a sample specific ‘STH code’ based on date of retrieval and operating list order whilst ensuring no clinical identifiers are present before transport to the research laboratory. A clinical-academic member of the team (OR) maintains the de-pseudonymisation key linking STH code to patient as an encrypted file within the Sheffield Teaching Hospitals IT infrastructure.
2.5. In the laboratory within a biological safety cabinet, transfer sample from the transport container to a 10 cm round tissue culture dish then gently rinse research specimen with PBS and aspirate off – repeat until clear.
2.6. Visually inspect the specimen to ensure tumour core (typically darker with evidence of necrosis) and adjacent infiltrated brain – ‘edge’ region (typically region most distant from tumour core with visible cortex, identified by sulcal/gyral patterning and/or visible grey-white differentiation) are present (see Figure 1B).
2.7. Using a scalpel, divide an approximately 5 mm × 5 mm sample from the ‘innermost’ region to represent the tumour core. Using a separate scalpel, divide a further 5 mm × 5 mm sample from the ‘outermost’ region to represent the infiltrative edge of the tumour. These samples represent resected (core) and typically residual (infiltrative edge) disease. Further 5 mm × 5 mm samples can then be dissected from the intervening regions using a further fresh scalpel to provide intermediate samples (Figure 1B). Separate each sample into an individually labelled culture dish.
Tip: We often take additional samples from both ‘core’ and ‘edge’ regions of an en bloc specimen to reflect spatial heterogeneity within each of these regions. We typically number samples (e.g. Core 1, Core 2, Edge 1, Edge 2) and find it helpful to take photographs demonstrating the macroscopic features of each sample prior to GSC derivation. Occasionally, we also take samples from tissue in the intervening region between ‘core’ and ‘edge’ extremities to generate ‘intermediate’ region GSCs.
3. Parallel derivation
Day 0
3.1. Using sharp dissection with a separate scalpel blade (those reserved above) for each sample, mechanically dissect the core, intermediate and edge samples aiming to produce fine pieces ≤1mm3 (see Figure 1C).
NOTE: We find cutting in one orientation using fast, repetitive up-down ‘chopping’ motion of the scalpel, then rotating the plate 90° and cutting again, then rotating a further 45° and cutting again provides fine, evenly divided pieces.
3.2. Add 5 ml of Accutase™ to each sample and pipette up and down using a separate glass or plastic Pasteur pipette for each sample to loosen large cell clumps, then incubate in a humidified cell culture incubator at 37°C, 5% CO2 and 21% O2 for 30 mins.
3.3. Examine each suspension under a microscope then pipette up and down again using a separate glass or plastic Pasteur pipette for each sample and return to the incubator for a further 15-30 mins.
3.4. Repeat step 3.3 until the tissue within each sample is predominantly dissociated and single cells are visible using a light microscope.
Tip: We generally find an overall incubation time of 45-90mins at 37°C provides optimal enzymatic dissociation, with exact timing influenced by a number of factors including: the size of each tumour sample; extent and homogeneity of preceding mechanical dissociation, and tumour consistency. Avoid excessive enzymatic dissociation as this can reduce cell viability leading to failure of the culture. We typically progress to step 3.5 as soon as roughly 50% or more of cells are visualised as single cells.
3.5. Using a fresh, sterile 10 ml serological pipette for each sample, transfer the cell-Accutase™ suspensions from the 10 cm dishes to labelled 50 ml Falcon tubes.
3.6. Add 5ml complete GSC media to each 10 cm dish, swirl to collect cells, then transfer this to the corresponding labelled 50 ml Falcon tube using a fresh serological pipette for each sample.
3.7. Filter each suspension through a 100 μm cell strainer rested on top of a second labelled 50 ml Falcon tube.
3.8. Centrifuge each cell suspension (core-filtered, intermediate-filtered, edge-filtered) at 1000 rpm (180 rcf) for 5 mins.
3.9. Very gently aspirate the supernatant whilst tilting each Falcon tube 45° then resuspend the cells in complete GSC media by gentle pipetting up and down 3-4 times using a serological pipette.
3.10. Transfer each cell suspension to a labelled T25 flask using a serological pipette and incubate upright in a humidified cell culture incubator at 37°C, 5% CO2 and 21% O2 for 37°C overnight.
Day 1
3.11. Warm complete media (3 ml per sample plus an additional 3-5 ml excess) in a water bath at 37°C then clean the container/bottle/falcon tube well with 70% IMS prior to use.
3.12. Transfer each GSC suspension (core, intermediate, edge) from old T25 flask to individual, labelled Matrigel™-coated T25 flasks using fresh serological pipettes for each flask and add a further 3ml of fresh complete GSC media.
3.13. Incubate with flasks lying on Matrigel™-coated side in a humidified cell culture incubator at 37°C, 5% CO2 and 21% O2 overnight to begin adherent monolayer formation.
Day 2
3.14. Warm complete media (5-7 ml per sample plus an additional 3-5ml excess) and PBS (5ml per sample plus excess) in a water bath at 37°C then clean the containers/bottles/falcon tubes well with 70% IMS prior to use.
3.15. Gently aspirate media off cells which will now be relatively adherent to the Matrigel™-coated flask using a serological pipette. Rinse very gently once with warmed PBS then aspirate off. Gently add 5-7 ml of fresh, complete GSC media then continue to incubate cells at 37°C, 5% CO2 and 21% O2.
Tip: We use appearances of the adherent cells under light microscopy prior to media exchange to guide the volume of media used. For example, using 5 ml complete media if very few adherent cells are observed (e.g. less than 10% flask surface area coverage/confluency) or 7 ml where adherent cells are more readily observed (e.g. greater than 40% flask surface area coverage/confluency)
4. Propagation
4.1. Maintain GSCs as an adherent monolayer (Figure 1D) in Matrigel™-coated flasks (initially in T25 flasks) by replacing complete GSC media 3x per week.
4.2. Once cells are 80-90% confluent, transfer cells from each T25 to a Matrigel™-coated T75 flask. This is performed by gently aspirating off old media using a serological pipette, rinsing very gently once with 5 ml warmed PBS which is aspirated off then adding 0.5 ml Accutase™ and incubating cells in a humidified cell culture incubator at 37°C, 5% CO2 and 21% O2 for 2-3 minutes to detach cells from the Matrigel™ coating. After this, 4.5ml of fresh, warmed complete GSC media is added to the T25 flask and the cell suspension is mixed by gently up-down pipetting 3-4 times using a serological pipette before transferring to a new T75 Matrigel™-coated flask. A further 5ml of warmed complete GSC media is added to the T75 flask before further gentle mixing of the cell suspension as above (3-4 times) and then incubation with flasks lying on the Matrigel™-coated side in a humidified cell culture incubator at 37°C, 5% CO2 and 21% O2.
4.3. Continue to expand GSCs by continued culture with regular media changes (3x per week) and splitting once cells are 70-80% confluent. Cryopreserve multiple vials of GSCs at the earliest available opportunity.
Tip: We advocate using patient-derived GSCs within experiments at the earliest passage possible – ideally within 10 passages, but typically less than 20. Nevertheless, we have been able to successfully propagate resected and residual GSCs for over 50 passages, demonstrating the feasibility of long term culture with this protocol, if required. At ultra-early passage (P1-3) we typically split heavy (i.e. 1:2-1:3) to reduce the risk of culture failure. After this stage splitting 1:4-1:10 is usually most appropriate.
4.4. Resected and residual model GSCs can also be cultured in the absence of Matrigel™ (or other adherent substrate) to form neurospheres or larger tumouroid cultures (see Figure 1D right – upper and lower panels).
4.5. To generate differentiated ‘bulk’ tumour cultures, during cell splitting a population of resected and/or residual model GSCs can be transferred to Matrigel™-coated flasks containing serum containing ‘bulk media’ (see Table 3). Performing experiments using both non-GSC enriched ‘bulk’ cultures and GSC cultures in parallel can be useful to establish differences in the biology and therapeutic resistance between these subpopulations.26,29,30
Tip: We propagate resected and residual model primary cells in bulk media for at least 5 passages before use in experiments as differentiated bulk cells. It is important when comparing resected and residual model GSCs to bulk cultures – to ensure these have been cultured in parallel and used in experiments at equivalent/near equivalent passage numbers. Additionally, we regularly use flow cytometry, immunoblotting and/or RT-qPCR to confirm reduced stem cell marker expression in differentiated bulk cells (see below).
| General guidance on suitability and inclusion/exclusion |
|---|
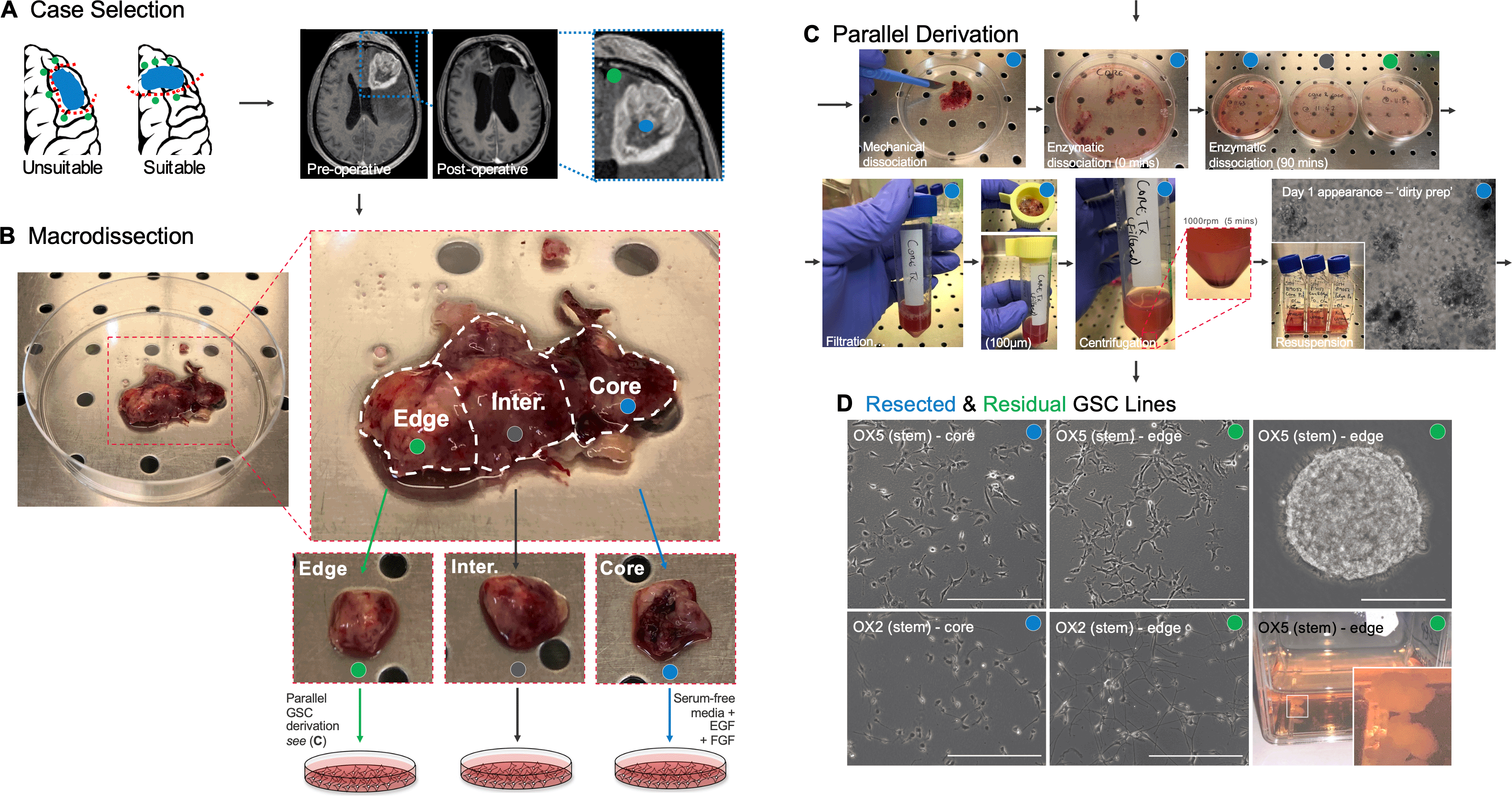
A. Selection of patients undergoing partial lobectomy for glioblastoma – schematic representation of unsuitable and suitable planned resections (left) and suitable pre- and post-operative MRI (T1-weighted) following intravenous contrast (right). Regions within the tumour core (for resected model) and adjacent brain tissue affected by the infiltrative edge (for residual model) highlighted in inset. B. En-bloc specimen following hemi-section in theatre for clinical pathology and research (left) before microdissection of the research specimen. Whilst maintaining orientation – the research specimen is subdivided as illustrated (right) to represent core, infiltrative edge, and intermediate (inter.) components for parallel GSC line derivation. C. Parallel derivation of patient-derived GSCs from core and edge of the en-bloc specimen – representative images. D. Representative brightfield light microscopy images (20x magnification) of Sheffield ex vivo resected and residual models. OX-5 and OX-2 core (left column) and edge (middle column) GSCs cultured as monolayers on Matrigel™-coated plasticware are demonstrated (scale bars = 500 μm). Right column (upper) – representative image of OX-5 edge GSCs cultured in non-adherent conditions as 3-dimensional neurospheres (scale bar = 250 μm), and; right column (lower) – representative photograph in non-adherent conditions (T25 flask) as larger 3-dimensional tumoroids following culture for 2 weeks. Scale bars = 200 μm.
Once successfully propagated, resected and residual cultures can be used within the range of common cell and molecular biology techniques to help better understand the biology of typically residual disease cells relative to those cells normally resected in a patient specific manner. Below, as a proof of concept, we apply common techniques to OX-5 core (resected) and edge (residual) cells – which is one of the first resected and residual models generated in our laboratory.
RT-qPCR was used to confirm whether OX-5 core and edge GSCs demonstrated elevated expression of key glioma stem cell markers relative to their bulk counterparts, and to explore any potential differences between the resected (core) and residual (edge) GSCs. OX-5 GSCs were seeded at a density of 25×105 cells per well into MatrigelTM-coated 6-well plates and incubated at 37°C in 5% CO2 for 48 hours in complete stem media without any treatments. RNA was extracted from primary cells using the Qiagen RNeasy Mini Kit according to the manufacturers protocol. Briefly, media was removed from cultured cells and cells were washed twice with ice cold PBS. PBS was aspirated and 350 μl RLT buffer was added to each well. Cells were dislodged using a cell scraper and transferred to labelled QIAshredder columns before being centrifuged at 8000r.c.f for minutes (4°C). Purple columns were discarded and 350 μl of 70% ethanol was added to the supernatant, which was then transferred to an Rneasy Minispin column and centrifuged for 15 seconds. Flow through was discarded and 700 μl of RW1 buffer was added to each column, which were then centrifuged for a further 15 seconds. This process was repeated twice with 500 μl of RPE buffer, with centrifugation steps of 15 seconds and then 2 minutes. Flow through was discarded at each step, then an additional centrifugation step for 1 minute was used to remove any excess ethanol. Following this, 50 μl Rnase free water per sample was added and these were centrifuged for 1 minute to elute the RNA. Total RNA levels in each sample were quantified using the NanoDrop ND 1000 Spectrophotometer (NanoDrop technologies); this quantifies absorbance a wavelength of 260 nm to establish the concentration of RNA in each sample, and the ratio of sample absorbance at 260/280 was used to confirm purity (absence of contaminants such as protein or phenol, which absorb strongly at/near 280 nm) with ratios ~2.0 deemed to represent ‘pure’ RNA.31 After RNA quantification, samples were stored at -80°C prior to use.
RNA samples were reverse transcribed using the High-Capacity RNA-to-DNA™ Kit (Applied Biosystems™) before the cDNA samples generated were used in triplicate reactions for each probe on a 384-well PCR plate (ThermoFisher Scientific). Each reaction consisted of: 2 μl cDNA, 5 μl TaqMan™ Universal PCR Mastermix (ThermoFisher Scientific), 2.5 μl ddH2O and 0.5 μl of probe (CD133 [probe assay ID: Hs01009259_m1], nestin [Hs04187831_g1], SOX2 [Hs01053049_s1], with GAPDH [Hs02758991_g1] used as a control ‘housekeeping’ gene; ThermoFisher Scientific). The plate was sealed using optical adhesive film (MicroAmp™, ThermoFisher Scientific) and loaded onto a 7900HT Fast Real-Time PCR System (ThermoFisher Scientific) to perform real-time quantitative PCR. The system was set to report 18S FAM with repeats of 40 cycles. Double delta Ct (2-ΔΔCt) analysis was used to determine relative gene expression using an average Ct value from the triplicate runs. At least 3 independent biological repeats of each experiment were performed using early passage (< P10) patient-derived cells of consecutive or near consecutive passage.
Immunoblotting was used to characterise any potential differences in the protein expression of key glioma stem cell markers between core (resected) and edge (residual) OX-5 GSCs. GSCs were seeded at a density of 25×105 cells per well into MatrigelTM-coated 6-well plates and incubated at 37°C in 5% CO2 for 48 hours in complete stem media without any treatments before media removal. Cell monolayers were washed twice in ice-cold (4°C) PBS then stored at -80°C. The plates were then thawed on ice before the addition of 100μl/well of lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM DTT and 1 mM EDTA supplemented with 50 U/μl benzonase (Novagen), protease and phosphatase inhibitors (Sigma)). Cells were harvested using a cell scraper and transferred to labelled eppendorfs on ice. Each sample was then vortexed for 5 seconds before incubation on ice for 15 minutes. The cells were then vortexed again for 5 seconds before an additional 15-minute incubation on ice and one final vortex. Cleared lysates were produced by centrifugation of the resulting samples at 15,000 rcf for 15 min at 4°C. Gel electrophoresis was performed using the NuPAGE system (Invitrogen).
Lysate (with LDS NuPAGE Sample Buffer, Invitrogen) volumes were calculated to provide 50 μg of protein loaded to each lane on the gels. SeeBluePlus2 Prestained Standard (Invitrogen) was loaded using 10μl alongside the protein samples as a molecular weight reference. Any empty wells were loaded with 5μl of 4x NuPAGE LDS Loading Buffer. The samples were electrophoresed at 140-150V for 90-120 minutes in an Xcell SureLock™ Electrophoresis Mini-Cell or Invitrogen Mini Gel Tank using 1x NuPAGE MOPS buffer (Invitrogen). Subsequently, protein within the gels was transferred to nitrocellulose membranes at 100V for 120 minutes in Mini PROTEAN Tetra Cells (BioRad) using 1x NuPAGE transfer buffer (Invitrogen).
Nitrocellulose membranes were then probed for the proteins of interest. All incubation steps took place on a rocking platform (approximately 30 rpm). Briefly, nitrocellulose membranes were blocked for 30 minutes at room temperature in 5% milk (Marvel, Tesco) dissolved in PBS-T. Meanwhile, primary antibodies were diluted to the appropriate concentrations (see below) in PBS containing 5% milk and 0.1% Tween-20 (Sigma). Membranes were then divided, based on the expected location of proteins of interest, then incubated with primary antibodies (CD133 1:1000 [Abcam ab216323 rabbit monoclonal, RRID:AB_2847920]; SOX2 1:500 [Santa Cruz sc-365823 mouse monoclonal, RRID:AB_10842165]; nestin 1:1000 [Abcam ab6142 mouse monoclonal, RRID:AB_305313]; β-tubulin 1:2000 [Abcam ab7792 mouse monoclonal, RRID:AB_306081]) overnight at 4°C on a rocking platform (at approximately 30 rpm) in the cold room. The next day, membranes were washed 3 times (10 minutes each + intervening washes) in PBS-T, before incubation with appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) at a concentration of 1:1000 (polyclonal goat anti-mouse IgG, HRP-linked [Dako P0447, RRID:AB_2617137]; polyclonal swine anti-rabbit IgG, HRP-linked [Dako P0399, RRID:AB_2617141]). Membranes were washed a further 3 times (10 minutes each + intervening washes) in PBS-T, then visualised using Pierce ECL western blotting substrate (ThermoFisher Scientific). In dark conditions, membranes were exposed to medical x-ray film (Fuji Medical Super RX, Fujifilm), then films were developed using a film processor (Mibolta SRX101A, Konica). Film images were then scanned to produce digital images using a scanner (Expression 1680 Pro, Epson).
Immunofluorescence was used to further visualise the relative protein expression of two key glioma stem cell markers (CD133 and nestin) within core (resected) and edge (residual) OX-5 GSCs. GSCs were seeded at a density of 5×105 cells per well in complete stem media onto MatrigelTM-coated coverslips inside a 24-well tissue culture plate. Cells were then incubated at 37°C in 5% CO2 and 21% O2 for 48 hours before all media was aspirated and 2 washes were performed with ice cold (4°C) PBS. The cells were then fixed using ice cold 4% PFA for 10 minutes followed by two washes with ice cold PBS. Cells were then permeabilised using 0.5% Triton X-100 (250 μl in 50 ml) and 3% BSA extraction buffer for 5 minutes at room temperature succeeded by 3 further washes with PBS. Primary antibodies were made up the required concentration in PBS with 3% BSA then 200μl of the required cancer stem cell marker antibodies (CD133 1:250 [Abcam ab216323 rabbit monoclonal, RRID:AB_2847920]; nestin 1:250 [Abcam ab6142 mouse monoclonal, RRID:AB_305313]) were added to each well and then plates were incubated overnight at 4°C in the refrigerator. The following morning the primary antibody was aspirated off before 3 additional gentle washes with PBS, then 200 μl per well of secondary antibody (Alexa Fluor® 594 Goat Anti-Mouse 1:1000 [Life Technologies A11005, RRID:AB_141372] and Alexa Fluor® 488 Goat Anti-Rabbit 1:1000 [Life Technologies A11034, RRID:AB_2576217]) diluted to the desired concentration in PBS with 3% BSA was added before wrapping all plates in foil and incubation at room temperature for 1 hour on a rocking platform (approximately 30 rpm). Following this incubation, the secondary antibody was aspirated off before 3 final washes (5 minutes each) in PBS (the second wash containing DAPI 1:1000) in dark conditions to prevent photo bleaching. Following the final wash, the cover slips were then mounted onto labelled microscope slides using Shandon Immu-Mount (Thermo Scientific). The slides were then stored in the dark to dry overnight.
Microscopy was performed on a Nikon Eclipse T200 inverted microscope and all images were taken using the Hamamatsu Orca ER camera and 200W mercury-xenon arc lamp at a total magnification of 600x (10x eyepiece, 60x objective). Images were captured using the Nikon NIS-Elements software package with image settings (including contrast, gain, offset) kept constant between experimental conditions. An excellent open access alterative to this software package would be ImageJ/Fiji. All images were converted to TIFF file formats and uploaded to the Collis laboratory Google Drive. Representative images are displayed.
Flow cytometry was used to quantitatively establish any potential difference in protein expression of the key glioma stem cell marker CD133 between core (resected) and edge (residual) OX-5 GSCs. GSCs were seeded at a density of 25×105 cells per well into MatrigelTM-coated 6-well plates and incubated at 37°C in 5% CO2 for 24 hours in complete stem media without any treatments. Media was aspirated off and cells were washed twice in PBS before 500 μl/well of Accutase™ was added. Both media and PBS washes were retained. Following detachment, cells were collected in 500 μl/well of PBS which was added to the media and PBS washes in 15 ml Falcon tubes labelled for each GSC line (OX-5 core and OX-5 edge). These were then centrifuged at 1,200 rpm (260 rcf) for 3 minutes. The supernatant was discarded, leaving a cell pellet to which 1ml of ice cold 70% ethanol was slowly added (whilst gently mixing on vortex for 5 seconds) to aid the fixation of cells in single cell suspension. Cells were left to fix overnight before excess ethanol was spun off and cells washed in PBS twice – centrifuging at 1,200 rpm (260 rcf) for 3 minutes and carefully discarding the supernatant at each stage. Subsequently cells were stained with Pre-conjugated mouse monoclonal [W6B3C1] antibody to CD133 (APC) for Flow Cytometry (CD133 1:10 [10 ul antibody in 100 ul cell suspension, approximately 100,000 cells]; catalog number A121882 antibodies.com [animal] monoclonal, RRID:AB_2876721) for 30 min at 4°C before flow cytometry was performed. Samples were processed using a LSRII flow cytometer (Becton Dickinson) with approximately 100,000 cells per sample and the data was analysed using FlowJo (Version 10, Becton Dickinson) software (open access alternatives include Flowing Software, FCSalyzer and, for those with more experience with R, the FlowCore and FlowJoWrappers R packages). Analysis of the FS and SS parameters was used to gate single cells, excluding both cellular debris and doublet cells. Additionally, a Red 633 laser was used to excite conjugated APC fluorophore, with the resultant emission detected in the red 660/20 channel. The fluorescence intensity of each cell detected was recorded and used to calculate the mean fluorescence intensity (MFI) for OX-5 core and OX-5 edge cells using FloJo analysis software. For each experiment, identical gating and analysis parameters were applied to each experimental condition to avoid bias.
Bulk RNA sequencing (RNAseq) was used to profile any further potential transcriptomic differences between resected and residual OX-5 GSCs at scale. GSCs were seeded at a density of 50×105 cells into Matrigel™-coated 10 cm dishes in complete stem media (see Table 5) and incubated for 48 hours at 37°C, 5% CO2, 21% O2 to allow for growth and adhesion. All media was then aspirated before 2 washes were completed with ice cold PBS and the resulting monolayers were then immediately frozen at -80°C until RNA extraction. Three biological replicates were prepared using cells at consecutive passages. RNA was extracted from core and edge GSCs using the Quick-DNA/RNA™ Miniprep Kit (Zymo Research) following the manufacturers protocol. Subsequently, a NanoDrop ND 1000 Spectrophotometer (NanoDrop Technologies) was used to quantify RNA and confirm sample purity (OD260/280 ratios >1.8). Samples were further analysed for RNA integrity using a Eukaryote Total RNA Nano-chip with the Agilent Bioanalyzer 2100 to ensure the samples met the parameters given by the RNA sequencing vendor Novogene (Total RNA extracts at concentrations of ≥ 0.8μg total RNA with extract concentrations of ≥ 20 ng/μl, with volumes of ≥ 20 μl or greater and RNA Integrity Number (RIN) of ≥ 6.8 or more per sample). Sample QC, library preparation and transcriptome sequencing was completed by Novogene (Cambridge, UK) with an aim of 10 million reads per-sample using an Illumina PE150. Raw sequencing reads (fastq) were processed using the nf-core RNA-seq pipline (nf-core/rnaseq) workflow - version 3.8.1. This includes pre-alignment quality control of counts and GC content via FastQC and identification, and removal of adapter sequences using Cutadapt.32 All sample reads were aligned to the reference genome Homo Sapiens GRC38 and transcript-level counts produced using the Salmon tool.33 The transcript-level were aggregated to the gene-level using the tximport R package.34 Principal component analysis (PCA) and differential expression analysis was conducted using the Bioconductor DESeq2 package35 within R Studio (Boston, MA) for estimation and visualisation of dispersion, Log2 Fold Changes, and -Log10 p-values.35,36 Heatmaps were generated using the pheatmap package in R.37 Volcano plots were generated with the tidyverse ggplot2 package.38 KEGG pathway enrichment analysis of significant genes (adjusted p-value < 0.05) was conducted using the clusterProfiler package39 and the KEGG Pathway database. Gene symbols were converted to Entrez IDs via the bitr function, and the enrichKEGG function was used to identify biological pathways significantly enriched. Outputs from the pathway analysis included count ratio, indicating the number of differentially expressed pathway genes as a proportion of the total number of genes in the pathway, and adjusted p-value (p-adjust).
Clonogenic studies were used to establish the feasibility of using OX-5 core and edge GSCs within these commonly used survival assays. GSCs were seeded in triplicate into Matrigel™-coated 6-well plates (2D clonogenics) or Matrigel™-coated Alvetex™ scaffolds in 12-well plates (3D clonogenics), which previous studies suggest may be more clinically predictive,26,40 at a density of 1,000 cells per well in 2 ml complete stem media. Plates were incubated at 37°C in 5% CO2 for 21 days prior to staining and counting. For 2D clonogenics, media was removed and colonies were stained using 0.4% methylene blue (Sigma-Aldrich) with a 60 minute incubation at room temperature before methylene blue was removed and any excess was washed from the plates by gentle submersion in tepid tap water. The colonies (greater than 50 cells) were counted. In contrast, 3D clonogenics were stained using 200 μL/well of MTT reagent (Sigma - 10 mg/ml in PBS) added to plates which were then incubated for 4 hours at 37°C in the dark. Importantly, in 3D clonogenic studies, MTT is used only to provide colony specific staining (since methylene blue can also discolour the Alvetex™ scaffolds) and the purple formazan product is not solubilised to estimate cellular activity or viability. Subsequently, media with MTT was aspirated off the scaffolds, plastic 12-well plate clips were removed, and 100 μL/well of 4% paraformaldehyde was added. Plates were incubated at room temperature for 15 minutes to allow fixation of the colonies, before removal of the excess paraformaldehyde. Plates with stained colonies can be foil wrapped and stored at 4°C for up to 14 days, but here were counted within 72 hours. The plating efficiency (PE) for clonogenic studies was calculated by the average number of colonies counted divided by the number of cells plated.
All in vitro experimental procedures consisted of at least 3 independent biological repeats (containing multiple replicates per group where specified). No formal power calculations were performed as this is a proof of concept, small-scale study and the applicability of the results is limited to the GSC model (and associated patient) in the study. If we were to conduct a larger study, formal sample size estimation would have been performed using a power calculation. Tests of normality (e.g. Shapiro-Wilk) were used to assess generated data sets prior to inferential statistics to confirm differences between test and control groups using appropriate statistical tests such as Mann-Whitney U-test (if normality was not apparent) or Student’s t-test (if normality was apparent) as specified in the results, with p-values <0.05 used to determine statistically significant biological differences while accounting for multiple testing where required. Inferential statistical analysis was performed using GraphPad Prism (Version 7), except where otherwise specified.
Here we present results generated using ‘OX-5 core’ GSCs and ‘OX-5 edge’ GSCs, some of the earliest resected and resected models within the SLB we have derived using the Sheffield Protocol detailed above, as a proof of concept. Since glioblastoma exhibit intractable therapeutic resistance and are universally fatal, we hypothesised that GSC lines generated from the infiltrative tumour edge within en bloc specimens to reflect post-surgical residual (normally 'left behind’) disease would demonstrate differences in mRNA expression of putative stem cell markers – since these are associated with resistance to radiotherapy,9 chemotherapy,41 and rapid tumour repopulation following treatment.10 If validated, this hypothesis would, at least in part, explain the failure of promising therapies evaluated in preclinical studies using glioblastoma cell lines generated from typically resected tumour cells to translate to improved patient survival in clinical trials, as the residual cells left behind after surgery (not the resected cells) are responsible for disease progression and death. Therefore, the question of whether resected cells and typically left behind (residual) cells are equivalent is of paramount translational importance.
Firstly, we assessed the mRNA expression of nestin in OX-5 core and OX-5 edge GSCs and matched bulk cultures (propagated in culture media containing serum for 5+ passages), alongside a more commonly used primary, patient-derived glioblastoma line (G1 GSCs and bulk cells).42,43 As expected, OX-5 core, OX-5 edge and G1 GSCs demonstrated greater nestin mRNA expression compared to matched bulk cell (non-GSC enriched) populations (p<0.0001) – see Figure 2A. In contrast, there was no demonstrable divergence in nestin mRNA expression between OX-5 edge GSCs and OX-5 core GSCs (p=0.9556). Similarly, there was no significant difference in nestin mRNA expression between OX-5 edge bulk and OX-5 core bulk cells (p=0.1070). Interestingly however, OX-5 core and OX-5 edge demonstrated higher nestin expression than G1 primary cells in both stem and bulk conditions. This may reflect the early passage nature of OX-5 cells used and/or interpatient variability, but nevertheless confirms expression of the stemness marker nestin in OX-5 core and edge compares well with a typical patient-derived GSC line used in recent studies.42,43
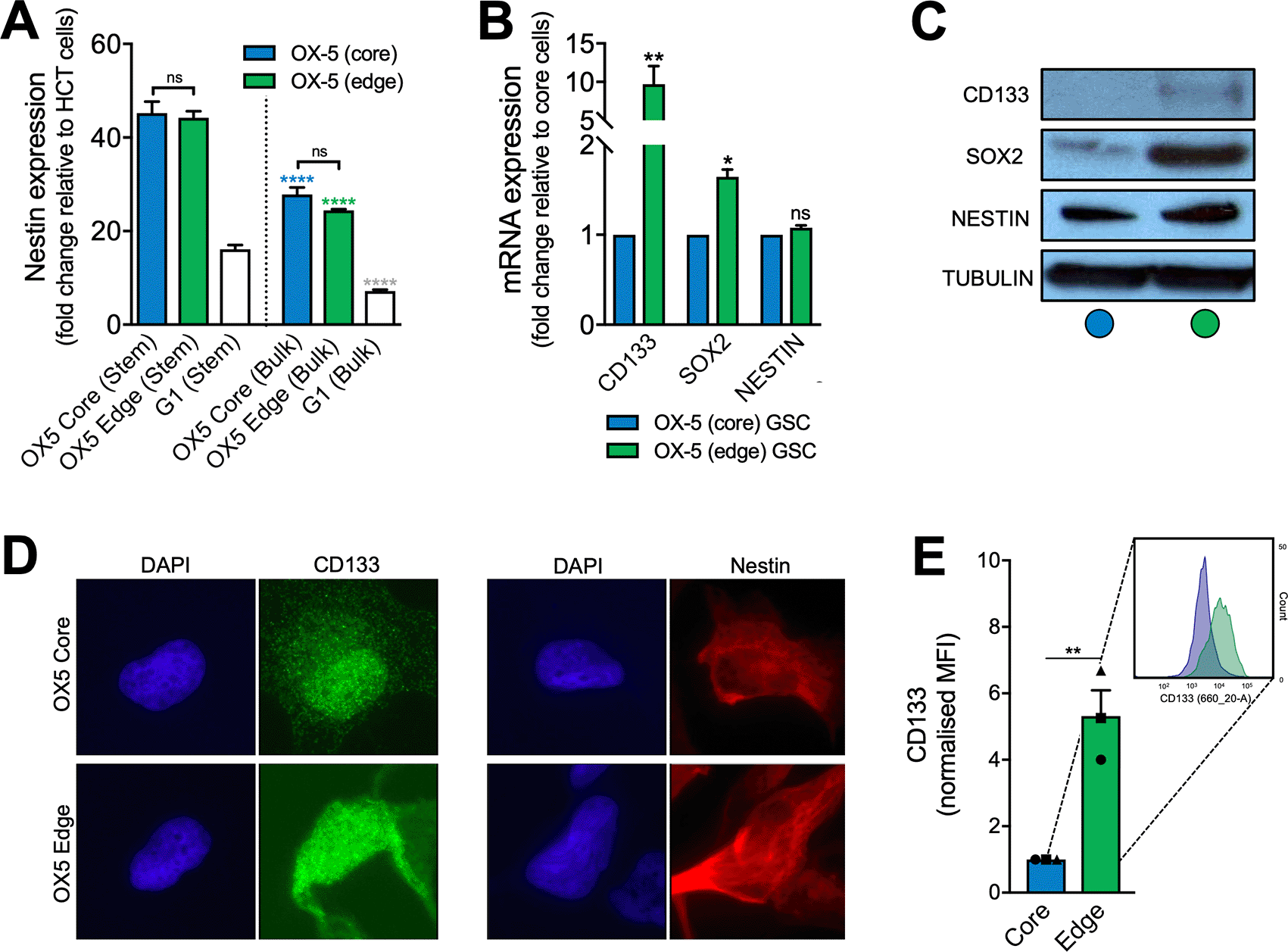
A. TaqMan™ RT-PCR analysis of nestin in untreated OX-5 core and OX-5 edge GSCs with matched bulk populations, to reflect resected and typically residual disease, respectively. These were evaluated for stem cell marker mRNA expression alongside G1 GSCs with matched bulk populations. Nestin expression normalised to HCT116 (colorectal cancer) cell line to facilitate comparison across cell lines and highlight elevated nestin expression across all the primary glioblastoma lines relative to an established non-glioma cell line. Asterisks (*/*/*) denote statistical significance of nestin expression in bulk cells compared to the corresponding GSC line. B. TaqMan™ RT-PCR analysis of CD133, SOX2 and nestin in untreated OX-5 core and OX-5 edge GSCs. Asterisks (*) denote statistical significance of stem cell marker mRNA expression in edge (residual model) stem cells compared to core (resected model) stem cells. In all experiments (A-B) cells were plated then incubated for 24 hours prior to RNA extraction. C. Immunoblot of glioblastoma stem cell marker expression in OX-5 core and OX-5 edge cells. D. Representative immunofluorescence images. E. Flow cytometric analysis of CD133 expression with representative histogram (inset). All graphs shown represent the mean of 3 independent experiments. Error bars represent the SEM.
Subsequently, we investigated expression of the additional commonly used glioblastoma stem cell markers CD133 and SOX2 in OX-5 GSCs, in addition to reassessing the expression of nestin. Validating the previous assessment, both OX-5 edge GSCs and OX-5 core GSCs demonstrated equivalent levels of nestin mRNA expression. However, in contrast, OX-5 edge GSCs demonstrated higher expression of SOX2 mRNA (1.6-fold higher, p=0.0274) and profoundly higher mRNA expression of the archetypal GSC marker CD13312 (9.7-fold higher, p=0.0033) relative to core GSCs from the same patient (Figure 2B). Subsequently, elevated SOX2 and CD133 expression in OX-5 edge GSCs was confirmed at the protein level using immunoblotting, immunofluorescence and flow cytometry (Figure 2C-E). Importantly, substantially higher CD133 protein expression (5.3-fold, p=0.0051) in OX-5 edge GSCs was demonstrated on flow cytometric analyses (Figure 2E), consistent with the profoundly elevated mRNA expression noted in these cells (above). These findings strongly support the possibility that infiltrative GSCs typically left behind after surgery may exhibit a distinct profile of ‘stemness’ and associated inherent therapeutic resistance compared to cells normally resected (and used in laboratory-based research). This hypothesis would be consistent with previous data demonstrating that GSCs demonstrated elevated potential for brain invasion44 and the landmark study by Bao et al. (2006)9 which demonstrated that GSCs, designated on the basis of CD133 positivity, displayed enhanced radioresistance associated with increased expression and activation of key DDR proteins including ATM, CHK2 and CHK1. Further detailed analysis of DDR capacity and activity within a wide range of residual and resected glioblastoma models within the SLB is ongoing in our laboratory and will be reported on in due course.
To elucidate more holistically whether these resected model and residual models generated in parallel from the same patient represent distinct tumour subpopulations, we characterised the transcriptomic landscapes of both OX-5 core vs edge GSCs using RNA-seq. Principal component analysis (PCA) of these data demonstrates that OX-5 core and edge GSCs differ vastly in the first principal component axis (PC1; Figure 3A). Furthermore, whereas OX-5 core GSCs exhibited a degree of transcriptional variability in principal component 2 (PC2) between independent biological repeats performed at consecutive passages (repeat 1 = passage 6; repeat 2 = passage 7; repeat 3 = passage 8), OX-5 edge GSCs clustered together, potentially indicating that residual (edge) GSCs may represent a more stable, consistent model to work with over time. However, it is important to emphasise that in multi-dimensional scaling the differences in PC2 are deemed of lower importance than differences identified in PC1. Therefore, the global transcriptomic divergence identified between OX-5 core and edge GSCs is more extensive than any divergence within either core or edge GSCs separated by time.
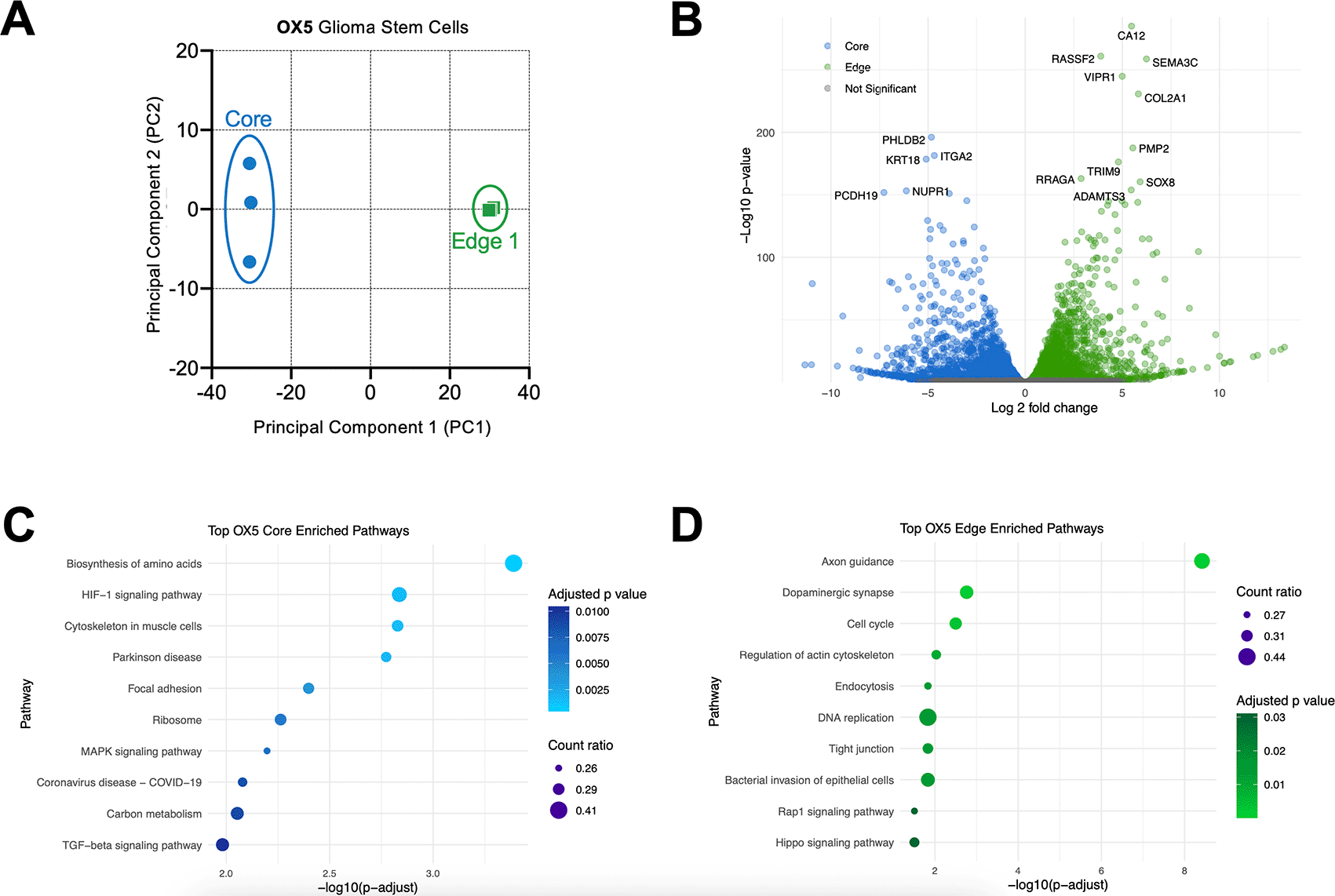
A. Principal component analysis (PCA) of RNAseq data from untreated OX-5 core and OX-5 edge GSCs, to reflect resected and typically residual disease, respectively. B. Volcano plot illustrating genes significantly (adjusted p-value <0.05) upregulated in resected (blue) and residual (green) disease GSCs. C-D. Pathway analysis to demonstrating top 10 KEGG pathways enriched in resected (blue) and residual (green) disease GSCs. All RNA seq data generated using 3 independent biological replicates/samples from GSCs at consecutive passages (for each independent biological sample there was one technical replicate i.e. each of the three samples per GSC line was sequenced once).
Further analysis of differentially expressed genes (DEGs) emphasised the magnitude of transcriptional divergence between these resected and residual models with 5,815 genes significantly more highly expressed in OX-5 core, whilst 1,773 genes were significantly more highly expressed in OX-5 edge. Genes with the most significant upregulation in OX-5 core GSCs included the transcriptional regulator nuclear protein (NUPR1), which roles include co-ordination of responses to cellular stress including oxidative stress45 and repression of ferroptosis,46 and genes with roles in shaping structural integrity and cell-to-cell interactions including PHLDB2, KRT18 (keratin 18), ITGA2 (integrin subunit alpha 2) and PCDH19 (protocadherin 19). In contrast, OX-5 edge GSCs demonstrated high expression of genes with implications for brain invasion and glioma-neuronal interactions including the matrix metalloproteinase ADAMTS, and SOX8, PMP2 and SEMA3C which have roles in differentiation towards astrocytic and oligodendrocytic lineages,47,48 axonal myelination,49 and axonal guidance,50 respectively (Figure 3B). These data were also consistent with gene ontology/pathway analyses with those most significantly enriched in OX-5 core GSCs including ‘biosynthesis of amino acids’ and ‘HIF-1 signalling pathway’, whilst those most significantly enriched in OX-5 edge GSCs included ‘axon guidance’ and ‘dopaminergic synapse’ pathways (Figure 3C-D). Collectively, these proof of concept data suggest resected (core) and residual (distant invasive edge) GSCs occupy highly distinct transcriptomic landscapes, which are likely to impact spatially-mediated functional heterogeneity and influence therapeutic response.
The clonogenic survival assay is recognised as a gold standard, clinically-relevant method for determining the sensitivity of cell cultures to candidate therapeutic agents including chemotherapy and ionising radiation. The assay measures viable cells that have demonstrated functional survival by maintaining the propensity for at least 5-6 rounds of replication to produce a colony.51,52 Interestingly, on assessment of clonogenicity, very early passage OX-5 core and OX-5 edge GSCs exhibited poor plating efficiency in 2D culture conditions despite extensive cell proliferation. This appeared largely due to an extremely migratory phenotype, with brightfield microscopy of 2D plates at the end of the colony formation period often demonstrating diffuse cell monolayers (data not shown). However, application of these resected and residual GSCs within a customised 3D Alvetex™ culture system, which as previously been validated to recapitulate key histological features of glioblastoma and better predict treatment response in clinical trials relative to traditional 2D cultures,26,27,40 demonstrated much improved clonogenicity (Figure 4) with a 3.4-fold increase in plating efficiency of 3D OX-5 core (p<0.0001) and a 2.1-fold increase in OX-5 edge GSCs (p=0.0008), relative to 2D conditions. Therefore, the use of resected and residual GSCs within the 3D Alvetex™ culture system may provide a robust and accessible platform to future studies to assess the influence of spatial intratumoural heterogeneity in differential responses to therapy in the preclinical setting.
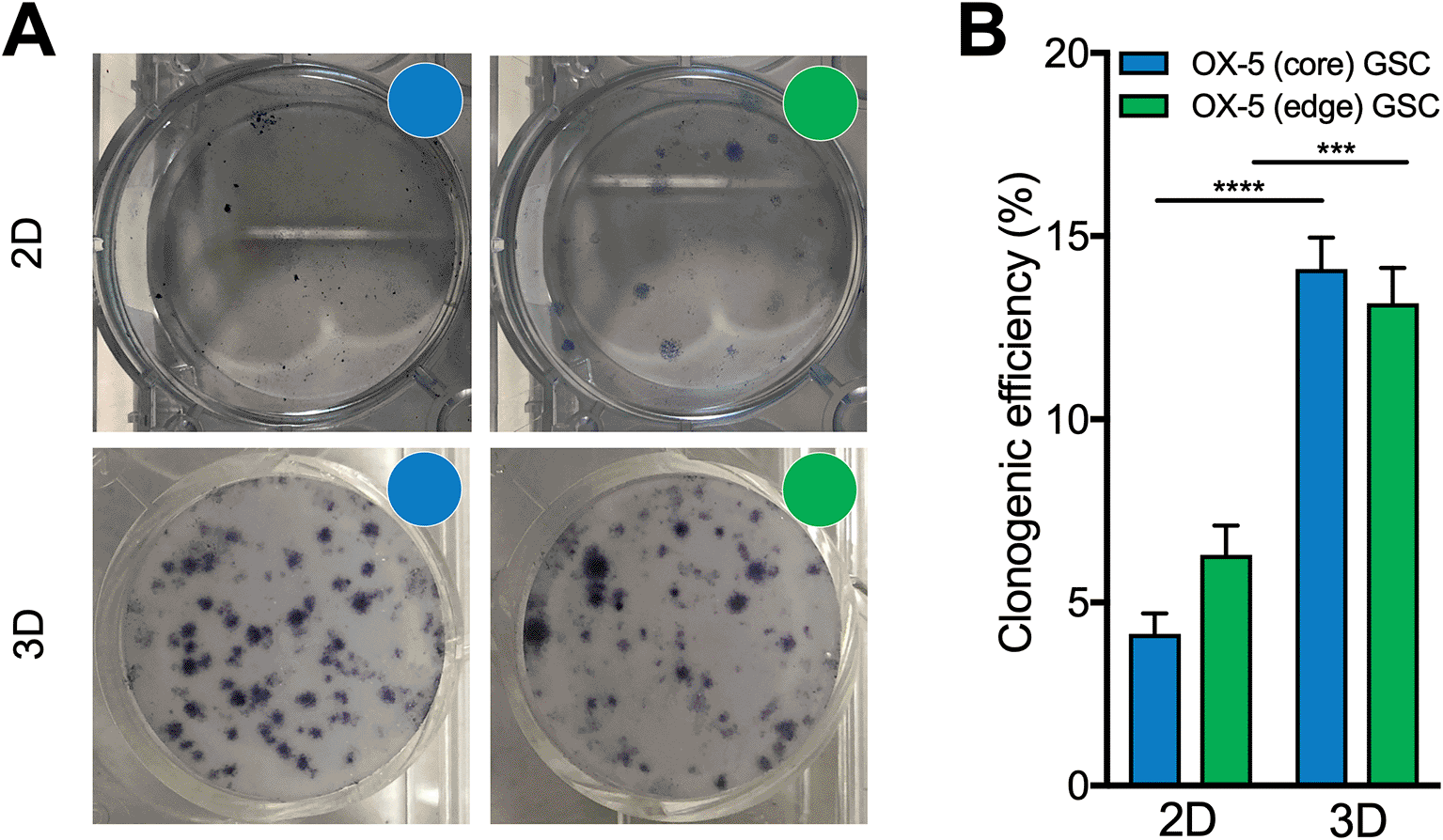
Untreated OX-5 core and OX-5 edge GSCs, to reflect resected and typically residual disease, were seeded at a density of 1,000 cells per well in 2D or 3D culture conditions (as indicated) then incubated for 21 days. Subsequently, colonies were identified using 0.04% methylene blue in 100% methanol to stain and fix 2D colonies, or incubation with MTT for 4 hours to reveal 3D colonies followed by paraformaldehyde fixation prior to quantification. A. Representative images, and; B. Comparison of clonogenic plating efficiency for OX-5 core and OX-5 edge GSCs in 2D and 3D Alvetex™ scaffold-based conditions. Data points shown represent the mean of 3 independent experiments (for each independent experiment there were 3 technical replicates for each condition). Error bars represent the SEM. Note 2D and 3D clonogenic plates not to scale – internal diameter of wells in 2D (6-well) plates is 2.3-fold the diameter of 3D Alvetex™ scaffolds (34.8 mm vs 15 mm).
Despite increasing evidence that intratumoural heterogeneity represents a major contributor to therapeutic resistance in glioblastoma,17–19 the availability of both in vitro and animal models systems that meaningfully recapitulate spatiofunctional heterogeneity present within patients in the early preclinical setting remains limited. Trends towards increasing use of serum-free protocols to derive and propagate patient-derived glioblastoma cells53–55 for over a decade have played a crucial role in advancing our understanding of inherently treatment resistant GSC subpopulations within tumours. However, it is becoming increasingly clear that not all GSC subpopulations are functionally equivalent56,57 and as such, there is a clear need to define anatomic or spatial variations in treatment response and/or resistance mechanisms. Indeed, an important study by Spiteri et al. (2018),18 which incorporated fluorescence-guided sampling of more distant infiltrative and subventricular zone glioblastoma cells, supports a hypothesis of genetically distinct tumour subclones that arise early in the evolution of glioblastoma; demonstrating a greater functional capacity to invade deeper into normal brain and therefore be more likely to represent disease left behind after surgery (residual disease).58,59 Since established cell and patient-derived orthotopic xenograft models commonly used in neuro-oncology research often fail to reflect the brain invasive phenotype of glioblastoma observed in patients,25,60 we have identified a previously unmet need. The replacement of some in vivo studies with the ex vivo residual disease models generated using tumour adjacent infiltrated brain tissue within en bloc tissue specimens from human patients may provide both scientific and 3Rs benefits.
The parallel ex vivo primary, patient-derived lines developed and presented here build on these prior studies by establishing models suitable to interrogate whether, and how, previously observed intra-tumoural spatial genetic heterogeneity18,61–63 will translate to functional heterogeneity, including intracellular responses to DNA damaging radiotherapy and temozolomide treatments (which represent standard-of-care adjuvant management for patients with glioblastoma) and subclonal divergence in therapeutic sensitivity. Crucially, post-surgical residual disease cannot be considered equivalent to glioblastoma at the time of recurrence, which may harbour a range of genomic changes following chemoradiotherapy.64 We demonstrate for the first time that distant infiltrative GSCs derived from tumour adjacent brain parenchyma within en bloc partial lobectomy specimens exhibit a distinct profile of key glioma stem cell marker mRNA expression (Figure 2), which may have implications for the responsiveness of residual GSC models to current therapy. These results are consistent with the study of glioblastoma tumour organoids by Hubert et al. (2016),65 which identified a higher proportion of SOX2+ cells at the rim of organoids relative to the core. However, our novel pragmatic methodology using rare en bloc partial lobectomy specimens provides the opportunity to interrogate the more distant infiltrative cells taken from patients and contrast these with GSCs from the tumour tissue normally resected in patient-matched way, and thus maximise the chance of defining quantitative differences and qualitative nuances between resected and typically residual disease. Developing a more detailed understanding of treatment naïve post-surgical residual disease, and how this may differ from resected disease (which forms the basis of current glioblastoma models), is likely to provide useful insights and inform more effective treatment of glioblastoma in the newly diagnosed setting.
Work by Smith & Rahman et al. using fluorescence-activated cell sorting of cells from resection margins sampled during 5-ALA fluorescence-guided surgery has provided an important approach to begin investigating the properties of cells more distant from the tumour core.66,67 However, spatially, these cells were based on the anatomical limits of a typical resection, and the isolation of ‘edge’ cells using a metabolism-based marker could potentially bias further investigation of complex divergence between core and edge cells. In contrast, the Sheffield Protocol (Figure 1) generates label-free ex vivo models of resected and post-surgical residual disease through a pragmatic approach which utilises relatively rare en bloc specimens (for example, where resection includes a partial lobectomy) to provide access to the most distant infiltrative cells feasible from a living patient without the need for any marker to select or enrich for a specific subpopulation of cells. These important specimens include portions of infiltrated brain that in most other circumstances would not otherwise be resected, and are large, which likely mitigates against a low proportion of cancerous (or 5-ALA fluorescing) cells at sites distant from the main tumour mass. We therefore suggest that GSCs derived from the most tumour-distant aspect of such samples are representative of bona fide residual disease and their propagation in parallel with GSCs derived from the tumour core can provide a powerful tool to interrogate differences in the complex biology of normally resected disease. These models are distinct from, and complementary to, recent models based on enriching for cells demonstrating 5-ALA induced fluorescence from tissue at the margins of a typical surgical resection.66,67 Considering that only the residual cells left behind after surgery are responsible for disease progression following standard-of-care therapy, developing a comprehensive understanding of residual disease is critical to help design future treatment strategies that account for spatial intratumoural heterogeneity and pre-empt disease recurrence.
From a 3Rs perspective, we firstly emphasise that the current translational paradigm for progressing potential therapeutic agents into the clinic typically through a series of in vitro studies using established and/or patient-derived cell cultures generated from the normally resected tumour mass, followed by in vivo studies, most commonly applying orthotopic PDX models through implantation of the same types of cell lines has not served patients with glioblastoma well. This is evidenced by the lack of new regulatory drug approvals since 2005 in the UK (temozolomide) and 2009 in the US (bevacizumab).68 Therefore, the generation and application of patient-derived resected (core) and residual (edge) GSCs cultured within both 2D and 3D models has the potential to replace some murine in vivo studies in high-grade glioma research, particularly in the context of efficacy studies testing drugs that have already progressed through clinical trials for other types of cancers and are therefore known to be clinically-safe/tolerable.
Furthermore, the patient-specific nature of each resected and residual GSC model generated could support replacement of animal studies specifically in the context of murine avatars for testing experimental therapeutics in glioblastoma. However, we do accept that confirmation of the clinically predictive nature of these models though assessing response to standard-of-care therapies in the patient-specific models and comparing this to actual response and survival amongst the associated patient cohort in the clinic would be important to provide definitive validation. This work is currently ongoing within our laboratory. Our ex vivo resected and post-surgical residual disease model advantages over subcutaneous and orthotopic PDX models also include speed of data generation (with robust survival data typically within 3 weeks using our 3D scaffold-based clonogenic assay) and scalability with the ability to test multiple drugs/combinations within a 12-well format and potential to integrate these models into 96-well 3D scaffold-based systems in the future.69 These advantages also highlight our model’s potential for reducing animal usage, as a range of candidate drugs and/or combinations could be effectively triaged prior to in vivo studies, resulting in fewer animals required to establish the most promising therapeutic strategy for clinical translation.
Therefore, given the prevalent use of animals in glioblastoma research globally, estimated to be over 75,600 animals/year based on our analysis of PubMed-indexed publications, we believe resected and residual GSC models have the potential to reduce and/or replace at least 5-10% of animal use (3,780-7,560 animals/year) in the contexts described above. To facilitate this, we are sharing our resected and residual models from within the SLB with researchers at an increasing number of academic institutions both nationally and internationally (see below), and we are actively engaging with our research ethics and Healthcare Gateway teams at the University and Royal Hallamshire Hospital to explore pathways that will enable the use of these models in studies conducted by Commercial partners (including Pharmaceutical Partners) to support residual disease informed drug discovery and evaluation.
As outlined in this methods paper, the Sheffield Protocol is based on macrodissection of suitable en bloc specimens and requires tight collaboration between clinical and academic members of the research team to ensure appropriate patient selection. Furthermore, a clear anatomical and spatial awareness follows each unique sample from the operating theatre to the bench. In this respect, we find the availability of a surgeon to stay with a sample from resection through to macrodissection into core, intermediate and edge subdivisions to be critically important. We believe that our protocol generates bona fide patient-derived core and infiltrative edge GSC lines in parallel, which exhibit divergent transcriptional landscapes (Figure 2), highly and differentially expressed putative glioblastoma stem cell markers (Figure 3), and can be easily applied to a range of assays and techniques commonly used to investigate cancer biology and assess novel therapeutic interventions, including clonogenic survival assays, immunoblotting, immunofluorescence, RT-qPCR and omic characterisation (Figures 2-4). The protocol is also simple to modify, for example, when a particularly large sample is provided, it is often feasible to generate multiple core and multiple edge GSC lines from the same en bloc specimen radiating out from the tumour core to distal invasive edge (where normal brain white matter can often be visualised). Conversely, when a sample is found to be unsuitable for resected and residual model derivation, we routinely generate between 3-5 parallel GSC lines from different tumour regions to generate ‘random intratumoural heterogeneity models’ which may provide useful additional controls in comparison to residual models in the future (i.e. establishing whether or not any specific divergent feature observed in residual disease models lies within the spectrum of variation observed portended by spatial heterogeneity within the core mass of the tumour).
Furthermore, in resections where the surgical team are able to record the precise retrieval location of some tumour tissue samples using intra-operative neuronavigation, MRI spatially-registered GSC models can be generated to further enhance spatiofunctional understanding. This approach can even be integrated with cases where resected and residual GSC models are generated following removal of the initial en bloc specimen, when the operating surgeon subsequently begins to resect further tumour infiltrated tissue from the posterior resection margin.
Using a combination of these approaches, our team has been able to generate the Sheffield Living Biobank (SLB) of patient-derived high-grade glioma. To date this includes over 150 models generated from 60+ patients (https://bit.ly/3yFF8WP) to help recapitulate the diverse repertoire of intratumoural and inter-patient heterogeneity observed in the clinic, and replace the need for animal studies in some contexts through the provision of more clinically-relevant cell culture models. SLB includes 44+ resected and residual GSC models and our team is delighted to share these with members of the neuro-oncology research community, as evidenced by our existing Materials Transfer Agreements with UK institutions for research use of SLB models (Figure 5).
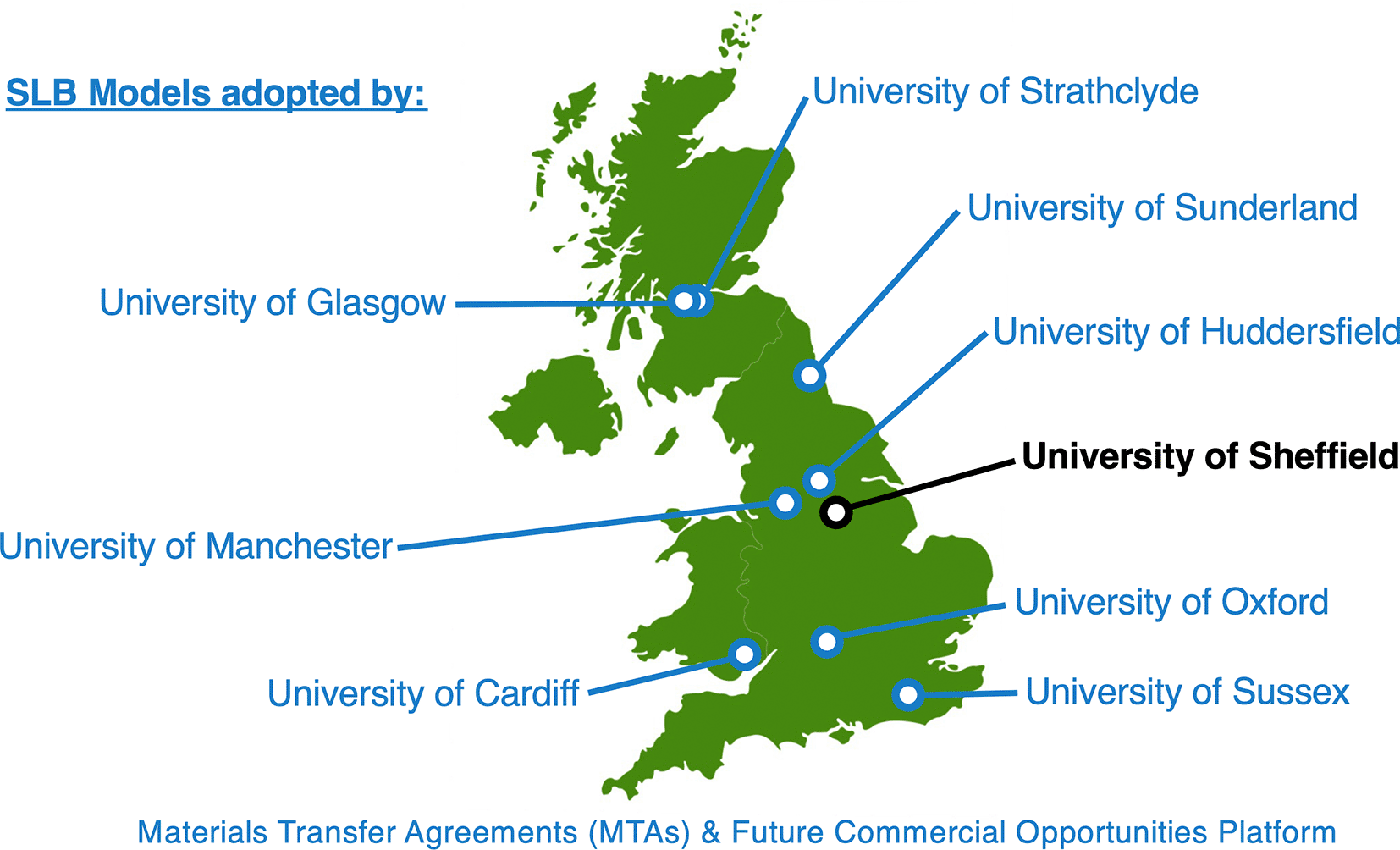
(UK map image credit: ELIKA/iStockPhoto).
MTAs have been established with 8 academic partner institutions to-date, with further potential MTAs in discussion with UK and international academic institutions (including in Sweden and South Africa). Work is also underway to facilitate use of these models within research completed by industrial partners. UK map image credit: ELIKA/iStockPhoto.
In conclusion, we present here a new robust methodology for the generation of surgically-relevant models of resected and residual glioblastoma that can be used to further elucidate the complex biology of these tumours, prevent unnecessary animal studies, and assess novel therapeutic interventions. We hope this can provide some much-needed and long overdue progress towards better treatment options for these patients.
The procurement of en bloc specimens suitable for resected and residual model generation requires appropriate institutional research ethics. At our institution, all clinical information and specimen acquisition was performed in within the scope of the approved IRB ethics protocol 11-YH-0319 (STH15598) - Yorkshire & The Humber - Leeds East Research Ethics Committee (approved 03/11/2011, substantial amendment approved 01/06/2016) and in adherence to the Declaration of Helsinki (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/). Close collaboration across academic and clinical boundaries with input from the neuro-oncology multi-disciplinary team (MDT) is essential.
Written informed consent for publication of the participants/patients’ anonymised associated clinical information and images was obtained from the patients providing brain tumour tissue for research under NHS REC approval STH15598.
Conceptualization: OR; SJC.
Data Curation: OR; CMG; KM; CGJ; KW; STW; MJD.
Formal Analysis: OR; CMG; KM; CGJ; KW; STW; MJD; SJC.
Funding Acquisition: OR; SJC.
Investigation: OR; CMG; KM; CGJ; KW; STW; AV; AGA; VB; MJD; AC; SS; DAJ; TAC; DW; YAT; SJC.
Methodology: OR; CMG; KM; CGJ; KW; STW; MJD.
Project Administration: OR; SJC.
Supervision: OR; NGR; AJC; YAT; SJC.
Validation: OR; CMG; KM; MJD; SJC.
Visualisation: OR; CMG; KM; KW; MJD.
Writing – Original Draft Preparation: OR; CMG; SJC.
Writing – Review & Editing: OR; CMG; KM; AV; VB; MJD; NGR; SS; DAJ; TAC; AJC; DW; YAT; SJC.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)