Keywords
pathogenic Staphylococcus, non-pathogenic Staphylococcus, open reading frames, comparative analysis, bacteria, pathogenicity
This article is included in the Genomics and Genetics gateway.
pathogenic Staphylococcus, non-pathogenic Staphylococcus, open reading frames, comparative analysis, bacteria, pathogenicity
The Staphylococcus genus consists of gram-positive cocci. The genus holds more than 40 species, grouped into pathogenic or non-pathogenic. Members of the pathogenic group are responsible for various infections such as nosocomial infections. However, non-pathogenic members are engaged in the food industry for the fermentation of cheese or meat. Scientists believe species habituate their pathogenic capabilities by possessing specific virulence factors acquired by horizontal gene transfer or mutations (Rosenstein & Götz, 2012).
Virulence factors encompass adhesins, exoenzymes, toxins, and a heterogeneous assortment. While adhesins interpose the attachment to host cells, exoenzymes destroy host tissue, and heterogeneous groups compromise iron uptake systems. Lastly, toxins directly exert damaging effects on the host. However, detecting any or all these factors in the staphylococcal genome does not make it pathogenic. For example, the non-pathogenic S. carnosus TM300 has the virulence factor sortase A (strA) within its genome. The strA is essential for mediating attachment to the host tissue, indicating that the role of strA is not exclusive and depends on the contribution of the cognate substrate proteins to the infectious pathway (Götz, Bannerman & Schleifer, 2006). Physiological properties significantly influence the pathogenicity of staphylococci. These properties impact their interactions with other pathogens, their ability to persist within the infected host, their resistance to antibiotics and antimicrobial compounds, as well as their capacity to evade neutrophil-mediated killing (Rosenstein & Götz, 2012).
Pathogenic staphylococci species’ ability to quickly adapt to antibiotic treatment is considered an indispensable feature. Antibiotic resistance genes, acquired by mobile genetic elements (transposons or plasmids), serve as mediators for this resistance ability, enabling rapid spread through lateral gene transfer or spontaneous mutation. The increasing resistance of staphylococci left only a few antibiotics effective in treating infections and increased species’ virulence ability (Ventola, 2015). For example, S. aureus has developed different strategies to counteract the effect of antibiotics resulting in the emergence of a new strain known as Methicillin-resistant Staphylococcus aureus (MRSA). MRSA alone is responsible for 11,285 deaths per year in the US, killing more Americans yearly than HIV, Parkinson’s disease, emphysema, and homicide combined (Guo et al., 2020).
A few studies have compared pathogenic and non-pathogenic staphylococci species. Such studies were usually limited to either the genomic aspect or performed on a small number of species. For instance, Rosenstein et al., (2009) analysed the genome of S. carnosus and compared a few features to S. aureus species. Heo and colleagues studied the genome of a few strains of S. epidiermidis, S. haemolyticus, and S. saprophyticus (Heo, Lee & Jeong, 2020). Although these species are opportunistic bacteria and involved in various infections, the study concluded that the genomes did not encode any virulence factors in S. aureus. Mannala et al. (2018) compared the genome of two highly virulent and low-virulent Staphylococcus aureus strains. Another study by Rosenstein & Götz (2012), defined the genomic information of pathogenic staphylococci species focusing on those derived from S. aureus strains. All previous studies highlighted similarities and dissimilarities between the genome of pathogenic and non-pathogenic staphylococci. Therefore, scrutiny of these analogies and diversity will enable us to understand the roots of their virulence ability and the followed infectious pathway.
The open reading frames are regions that either contain no stop codons or begin with a start codon and end with a termination codon. Each strand of the DNA sequence has three possible reading frames. Exploring bacterial ORFs provides an opportunity to discover novel functional genes (Cerqueira & Vasconcelos, 2020). Recently, biologists have been more concerned about the small ORFs (smORFs) (<50 amino acids) that manifest a vital role in several cellular regulatory activities, and more studies have focused on developing new approaches to annotate them (Mir et al., 2012). Intrinsic and extrinsic are the two in-silico methods for detecting ORFs. While the intrinsic pathway investigates ORFs coding potential, such as obvious ribosome binding site (RBS), the extrinsic technique hunts for conserved sequences among different species (Cerqueira & Vasconcelos, 2020). The latter is a potent approach for detecting smORFs (Warren et al., 2010; Wood et al., 2012; Cerqueira & Vasconcelos, 2020), considering that confirmed short-protein coding genes are without any marked RBS (Hemm et al., 2008). However, as of now, the number of annotated smORFs is considerably low.
In this study, we comparatively analysed ORFs extracted from ten selected staphylococci species, including five pathogenic and five non-pathogenic strains. Our objectives were to characterize the features of ORFs in pathogenic genomes, identify conserved ORFs specific to pathogenic staphylococci, and propose a novel approach for smORFs’ prediction and annotation. This study holds significance in addressing the need for comparative investigations of ORFs in pathogenic and non-pathogenic staphylococci genomes and contributes to the growing attention towards smORFs.
GenBank (Benson et al., 2010) and RefSeq (O’Leary et al., 2016) databases contain genome sequences of 98,079 staphylococci strains. We selected species whose pathogenicity is defined and confirmed for the comparative analysis. S. aureus Mu3 (Refseq assembly accession ID: GCA_000010445.1), S. lugdunensis HKU09-01 (GCA_000025085.1), S. haemolyticus JCSC1435 (GCA_000009865.1), S. saprophyticus ATCC 15305 (GCA_000010125.1), and S. schleiferi strain 1360-13 (GCA_001188855.1) represented the pathogenic species, while the non-pathogenic were S. carnosus TM300 (GCA_000009405.1), S. cohnii SNUDS-2 (GCA_001990205.1), S. warneri SG1 (GCA_000332735.1), S. nepalensis DSM 15150 (GCA_002902745.1) and S. pasteuri JS7 (GCA_002442915.1). We downloaded the genome sequence of these selected genomes from the NCBI FTP site (https://ftp.ncbi.nlm.nih.gov).
We employed version 6.6.0.0 of the EMBOSS getorf algorithm to extract open reading frames (ORFs) from the genomes specified in the previous section (Rice, Longden & Bleasby, 2000). The process involved running a Linux script with specific parameters for the EMBOSS getorf algorithm: ‘getorf -sequence genome.fa -find 1 -outseq genome_orf.txt.’ The optional qualifier -find [1] was utilized to determine the translation of ORF regions between the start and stop codons.
The identified ORFs were arranged according to size and any ORF shorter than 10 amino acids (aa) were discarded. Next, we identified shared ORFs within and between the two species groups. Eventually, we categorized these common ORFs into five groups based on their presence in the tested genomes: ORFs present in all tested genomes, ORFs present in all pathogenic genomes, ORFs present in all non-pathogenic genomes, ORFs present in some pathogenic genomes, and ORFs present in some non-pathogenic genomes (Underlying data: Appendix A–E) (Farhan et al., 2023). Figure 1 visualizes the ORFs filtration process (Farhan, 2023).

The functional annotation of ORFs followed two approaches: (i) a direct approach that utilized the annotated proteins files available in GenBank and RefSeq databases and (ii) an indirect approach based on the traditional BLASTp tool version 2.11.0 (Altschul et al., 1997). Following the direct approach, the sequence and coordinate of each ORF matched with its resembled annotated protein of the tested genomes — ORFs which failed in the direct annotation tested for the indirect annotation that utilized the BLASTp tool. The BLASTp tool parameters were adjusted to search in the non-redundant protein sequence database for homologous sequences to ORFs only in the Staphylococcus organism (taxid 1279), targeting a maximum of 100 species. Both identity level and query coverage should be higher than 85%.
The Blast2GO tool version 5.2.5 (Conesa et al., 2005) enables an efficient automatic functional annotation of protein sequences according to the gene ontology vocabulary. Gene ontology (GO) describes the biological framework of genes in three aspects: biological process, molecular function, and cellular component. The relationship between GO terms, when presented in graph-based terminology, the parent GO terms, refers to the node closer to the roots (Level = 2) of the graph and a child (Level ≥ 5) to that closer to the leaf nodes. Moreover, the algorithm performed the gene enrichment analysis and two-tailed Fisher exact test to identify the enriched biological processes in pathogenic tested genomes.
The DeepGOPlus algorithm (Version 1.0.2) was operated to predict the ORFs function, where their function was previously unknown (Kulmanov, Khan & Hoehndorf, 2018). The algorithm uses deep learning to learn features from query protein sequences besides cross-species protein-protein interaction networks. The resulting output was in the structure of GO terms, and the terms were presented in a graph chart using the QuickGO tool version 1.15 (Binns et al., 2009).
A conserved sequence is an amino acid sequence in a protein (or a nucleotide base in DNA) that has remained unchanged throughout evolution to maintain a protein’s structure and function (Alberts et al., 2002). Testing the conservation level of unknown ORFs (unORFs) involved three stages: conservation within the Staphylococcus genus, conservation within pathogenic staphylococci species, and finally, extending the analysis beyond the Staphylococcus genus. We employed the tblastn algorithm version +2.11.0 (Altschul et al., 1997) to conduct the conservation test, gathering data based on identity level and query coverage (≥85). The student t-test provided by Python’s SciPy library was employed to assess the results’ significance.
Clustal Omega – a multiple sequence alignment (MSA) algorithm version 1.2.4 was used to align the sequences to assess the locus conservation assay (Sievers et al., 2011).
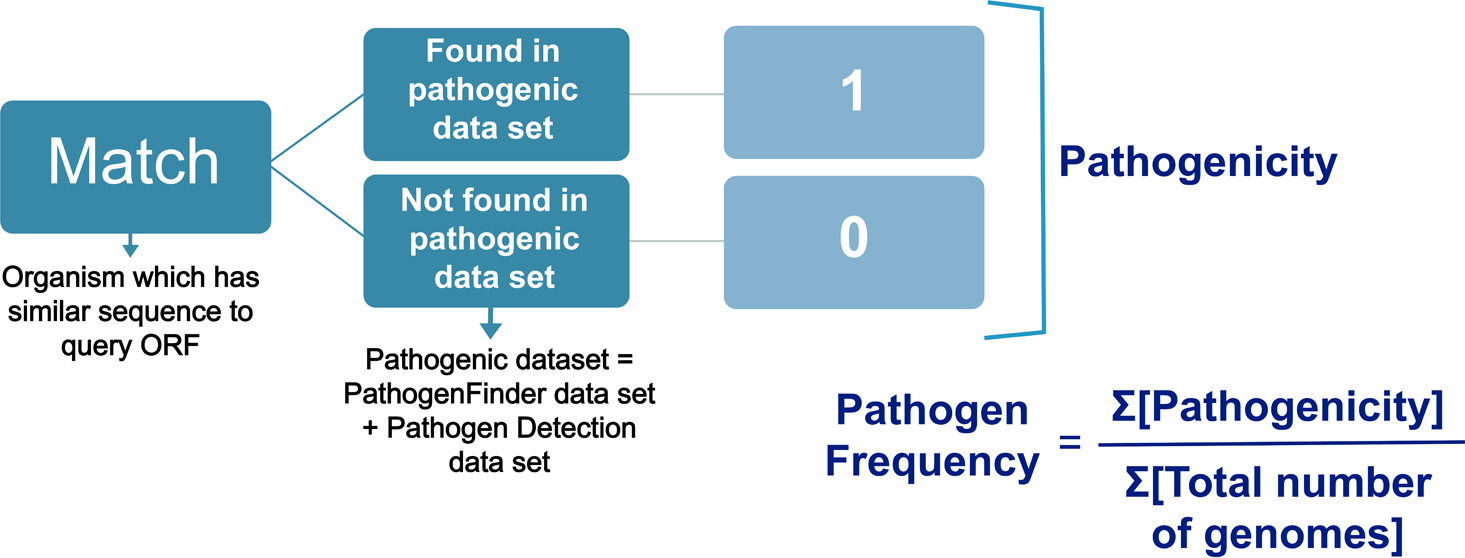
Both PathogenFinder 1.1 and NCBI Pathogen detection datasets (Cosentino et al., 2013; NCBI, 1988) facilitated entitling the species as either known to be pathogenic (1) or unknown pathogenicity (0). An ORF’s pathogenicity refers to the number of pathogenic genomes that own a homologous sequence. The pathogen frequency of an ORF was specified by dividing the pathogenicity of an ORF by the total number of genomes (Figure 2).
The Prodigal (Prokaryotic gene recognition and translation initiation site identification) algorithm (Version 2.6.3) pinpointed the ribosomal binding site (RBS) motifs in the S. aureus Mu3 genome (Hyatt et al., 2010) to verify whether the RBS motif preceded the ORF or not.
To explore the neighbouring genes for each ORF and outline its precise locus, the interval between all selected ORFs (selORFs) and genes of the S. aureus Mu3 genome was measured per their coordinates. We downloaded the annotated protein file for S. aureus Mu3 from Genbank FTP website.
The analysis started with extracting ORFs from 10 selected genomes (five pathogenic and five non-pathogenic) and any ORF smaller than 10 amino acids was excluded from the analysis. Subsequently, we categorized them into five groups based on their presence in the tested genomes (Table 1). The results revealed that six ORFs were common to all tested genomes across both groups. Among the found in some pathogenic tested genomes group, 1572 ORFs were present, and 15 unique ORFs were specific to all pathogenic genomes. Likewise, some non-pathogenic genomes exhibited 1567 identified ORFs; all non-pathogenic genomes contained 13 exclusively unique ORFs.
Table 2 summarizes the results obtained from ORFs annotation via direct and indirect approaches. Interestingly, one ORF was identical to a part of the 30S ribosomal protein S9 sequence of all non-pathogenic tested genomes.
Our methodology provided information about several hypothetical proteins whose functions were unknown. For example, the hypothetical protein in S. lugdunesis HKU09-01 overlapped with cell surface protein IsdA, indicating a role in transferring heme from haemoglobin to apo-IsdC. In S. aureus Mu3, the glycerophosphoryl diester phosphodiesterase homolog protein was identical to the unnamed protein product in S. haemolyticus JCSC1435. Parallel to results obtained from annotating ORFs of pathogenic tested genomes, the hypothetical protein of S. cohnii SNUDS-2 was identical to the YlbF/YmcA family competence regulator protein of S. nepalensis DSM 15150. In a nutshell, we detected 15 hypothetical proteins identical to known functional proteins shared between either some non-pathogenic or pathogenic species.
Both pathogenic and non-pathogenic groups have similar functional proteins, such as the 50S and 30S ribosomal proteins, translation initiation factors, acyl carrier proteins, ATP-binding proteins, transposase, and transcriptional regulators. Although both groups have 50S and 30S ribosomal proteins and transposase, they differ in counts and types. Eleven ORFs of pathogenic genomes overlapped with 28 proteins of either 30S or 50S ribosomal proteins, whereas 31 ORFs of non-pathogenic genomes overlapped with 65 ribosomal proteins. Sixty-one transposases were detected in pathogenic genomes, while only eight transposases in non-pathogenic genomes. The only family type familiar to both groups was the IS256 transposase family. According to the annotation results, 35 functions were unique to pathogenic genomes. In contrast, 38 processes were exclusive to non-pathogenic staphylococci.
In the context of exploring biological process GO terms (GO:0008150), our investigation revealed that multiple ORFs within both pathogenic and non-pathogenic tested genomes were associated with cellular process (GO:0009987) and metabolic process (GO:0008152) terms; however, they had several differences. The biological regulation (GO:0065007) GO term was dominant in the non-pathogenic compared to the pathogenic group (Figure 3A). The localization (GO:0051179), DNA integration (GO:0015074), DNA recombination (GO:0006310), cation transport (GO:0006812), and metal ion transport (GO:0030001) GO terms mapped solely to some ORFs from pathogenic tested genomes. In contrast, eight GO terms were exceptional to ORFs of non-pathogenic genomes (Figure 3B).
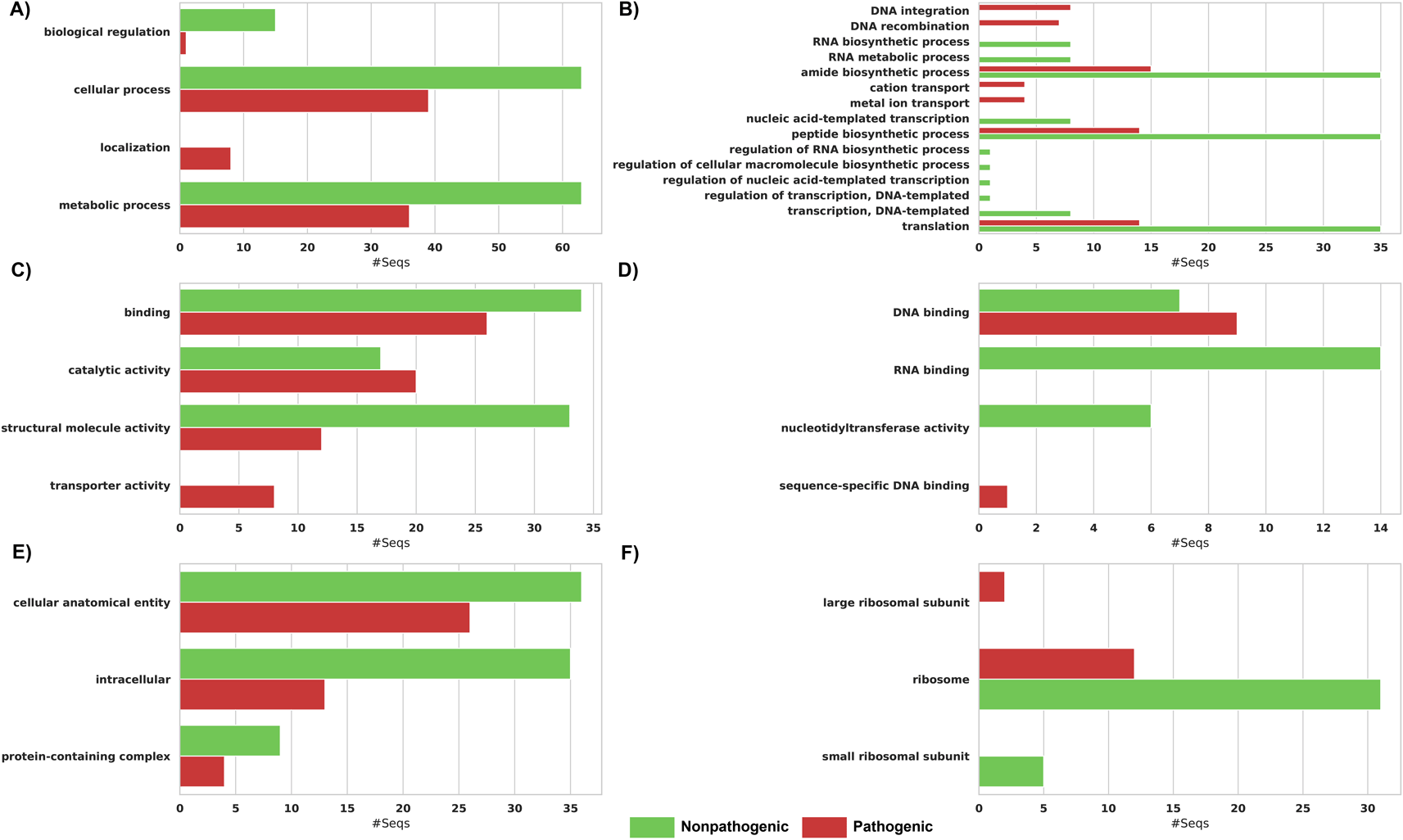
(A) Biological process parent's GO terms. (B) Biological process child's GO terms. (C) Molecular function parent's GO terms. (D) Molecular function child's GO terms. (E) Cellular component parent's GO terms. (F) Cellular component child's GO terms.
Concerning the molecular function (GO:0003674) GO term’s map, the transporter activity (GO:0005215) and sequence-specific DNA binding (GO:0043565) were exclusively associated with several ORFs of pathogenic tested genomes (Figure 3C). On the other hand, nucleotidyltransferase activity (GO:0016779) and RNA binding (GO:0003723) GO terms were unique to ORFs of non-pathogenic genomes (Figure 3D).
When comparing the cellular process (GO:0005575) GO terms (Figure 3E), ORFs of pathogenic tested genomes were exclusively associated with the child GO term, large ribosomal subunit (GO:0015934) (Figure 3F). In contrast, ORFs of non-pathogenic tested genomes displayed specificity, aligning solely with the child GO term, small ribosomal subunit (GO:0015935).
In the enrichment analysis, biosynthetic (GO:0009058), cellular biosynthetic (GO:0044249), and organic substance biosynthetic (GO:1901576) processes were under-represented in the pathogenic compared to the non-pathogenic group with equal significant p-value (3.12E-05) and False Discovery Rate (FDR) value (0.0046016).
Ultimately, several functional characteristics distinguished ORFs of pathogenic tested genomes compared to ORFs of non-pathogenic genomes, and vice versa. We were left with many ORFs from both pathogenic and non-pathogenic species whose functions were unknown. These ORFs did not overlap with any annotated proteins, hits from the BLAST search were below the specified threshold, and they were not annotated to any GO term by the Blast2GO tool. We referred to these as unknown ORFs (unORFs).
The analysis revealed significant similarity between 816 unORFs from pathogenic tested genomes and over 49,000 sequences in various staphylococci species. This similarity was notably higher than observed among the 810 unORFs from non-pathogenic tested genomes, with a p-value of 5.59e-16 tested by U-Mann Whitney test, indicating substantial conservation (Figure 4A). Moreover, the identified unORFs from pathogenic genomes exhibited sensitivity to pathogenicity (p-value: 2.99e-43) (Figure 4B). Among these, 23 unORFs demonstrated exceptionally high conservation within pathogenic staphylococci genomes, displaying a pathogen frequency ≥ 0.98. We designated these as selected ORFs (selORFs). These selORFs, with an average size of 21 amino acids, exhibited specificity toward pathogenic staphylococci species (Underlying data: Appendix F) (Farhan et al., 2023).

(A) Conservation of unORFs in pathogenic and non-pathogenic genomes within staphylococci species. (B) UnORFs of pathogenic tested genomes conservation within pathogenic and unknown pathogenicity staphylococci species. (C) Selected ORFs (selORFs) conservation outside the Staphylococcus genus.
Subsequently, we explored the conservation level of the 23 selORFs beyond the Staphylococcus genus. Among them, selORF with ID AP009324.1_34709 and four other selORFs emerged in 293 genomes outside the staphylococci species (Figure 4C). Notably, most of these genomes (208 out of 293) belonged to the Bacillus genus.
Gene ontology and gene product
DeepGoPlus predicted that our selORFs play a role in the mRNA catabolic process (GO:0006402) besides sharing functional similarities with the Pelota gene. However, the predicted GO term’s confidence level was between 0.3 and 0.4, considering algorithms find it challenging to find patterns in short sequences.
Further, the Prodigal algorithm indicated that neither of the selORFs was downstream of an RBS motif. Hence, we explored the neighbouring genes to test the hypothesis that our selORFs were likely non-coding RNA, translated on different frames, and probably engaged in regulatory functions.
Neighbouring genes and anti-sense sequence
Regulatory small proteins regulate their neighbouring genes or genes on the opposite strand. We measured the interval between selORFs and genes within the model genome (S. aureus Mu3). The mean distance between selORFs and genes on the forward strand was 19.203 (log2), a value comparable to the mean distance of selORFs on the reverse strand 19.0951 (log2). Based on these distances, we categorized the selORFs into two groups: (i) those with a zero distance and (ii) those with a non-zero distance. In the first category, selORFs exhibited overlaps with other genes on the same or opposite strands, occurring in various reading frames.
Nine selORFs demonstrated overlap with genes positioned on the same strand. Among these, five selORFs exhibited overlap with coding genes, including transposase, hypothetical protein, and serine protease genes. Conversely, four selORFs displayed overlap with non-coding rRNA genes, as indicated in Figure 5A. Interestingly, two distinct selORFs showed overlap with the same rRNA gene (SAHV_r0002), mirroring a similar occurrence with the transposase gene (SAHV_2363). Notably, the extent of overlap remained constrained, with the selORFs covering at most 13% of the gene size.

(A) Distribution of SelORFs overlapped with genes within the model genome (S. aureus Mu3). (B) selORF to gene size ratio distribution.
A total of 10 selORFs displayed overlap with genes situated on the opposite strand, with seven of these being coding genes and the remaining three being non-coding genes (Figure 5B). Within this set of 10 selORFs, eight exhibited overlaps with segments of individual genes on the opposite strand. Among these, three selORFs overlapped with tRNA-Val and rRNA non-coding genes. The remaining five selORFs demonstrated overlap with specific genes, namely hsdM (BAF77312.1), 50S ribosomal protein L1 (BAF77419.1), hypothetical protein (BAF77875.1), graD (BAF78358.1), and type I restriction enzyme EcoR124II M protein homolog (BAF78676.1). Furthermore, two selORFs displayed overlap with the 5′ and 3′ ends of distinct genes located on the opposite strand.
When the distance between a selORF and a gene was not zero in the second group, the selORFs did not overlap with any genes within the tested genome. In total, seven selORFs fell into this group. Among these seven, only three displayed notable proximity to genes, with distances less than 5 log2 units. Specifically, selORFs identified by the IDs AP009324.1_3643 and AP009324.1_3911 were close to the rRNA genes on the same strand (SAHV_r0003 and SAHV_r0007, respectively). In contrast, the selORF AP009324.1_34650 was near the 30s ribosomal protein S12 gene (SAHV_0543) on the opposite strand.
Of particular interest is the selORF previously mentioned (ID: AP009324.1_34709), which exhibited remarkable conservation in 102 of 200 pathogenic species. Most notably, this selORF stood out as the sole instance present in 100 non-Staphylococcus genomes, and it overlapped with the 50S ribosomal protein L1 on the opposite strand across these 200 genomes.
Notably, the termination codon of selORF was positioned 305 nucleotides away from the start codon of the former ribosomal protein in 10 genomes, signifying a conserved genetic location for selORF. According to MSA, the amino acid sequence shared with our selORF within the 50S ribosomal protein L1 exhibited 100% identity across the adopted genomes. However, variations were observed in nucleotide sequences, attributed to differing frame translations. Aligning the investigated protein in each adopted genome yielded varying similarity scores compared to the S. aureus Mu3 50S ribosomal L1 protein (Figure 6A).

Furthermore, we examined the specific selORF within genomes closely related to the Staphylococcus genus. According to NCBI taxonomy (Schoch, 2011), genomes such as Salinicoccus alkaliphilus DSM 16010 (NZ_FRCF01000009.1), Salinicoccus albus DSM 19776 strain YIM-Y21 (NZ_ARQJ01000028.1), Salinicoccus carnicancri Crm 50.SCCRM.1_10 (NZ_ANAM01000010.1), Nosocomiicoccus ampullae strain DSM 19163 (NZ_JACHHF010000004.1), and Nosocomiicoccus massiliensis isolate MGYG-HGUT-01449 (NZ_CABKSY010000018.1) were identified as closely related to the Staphylococcus genus. Interestingly, none of the 50S ribosomal protein L1 sequences in these analogous genomes matched the corresponding protein sequence in S. aureus Mu3 (Figure 6B). However, the MSA algorithm displayed that the region encompassing selORF exhibited similarity in amino acid and nucleotide sequences across all related genomes.
This selORF displayed significant conservation, particularly concerning the well-preserved 50S ribosomal protein L1 across multiple species. Despite its relatively small size (18 amino acids), the possibility of obtaining a functional protein remained notable within the Staphylococcus genus (0.0002964), as well as in Bacillus (0.0023744), all bacteria (0.00083889), and all organisms (0.0056284), based on data from the UniProt database.
Both pathogenic and non-pathogenic staphylococci species occupy conserved proteins responsible for translation, replication, and survival (Rosenstein et al., 2009). Even though our results showed that both groups share the same fundamental functional proteins, each group developed genes that facilitate specific functions according to their adopted lifestyle. Results captured from the comparative analysis manifested their significance in various means by identifying functions of 15 hypothetical proteins, providing hints of the functional characteristics of each group, and highlighting a new methodology for spotting smORFs.
At this point, what distinguishes one group from another is still obscure, as there is a lack of studies comparing pathogenic to non-pathogenic species. Staphylococci species that are generally recognized as safe are known to be associated with food fermentation. Previous studies observed increased antioxidant activities in fermentation (Barrière, Leroy-Sétrin & Talon, 2001; Abubakar et al., 2012). Thiol reductase thioredoxin, oxidoreductase, and cytochrome aa3 quinol oxidase (restricted to non-pathogenic tested genomes) are enzymes required for the antioxidant pathway in bacteria. This feature of non-pathogenic species suggests it has been acquired as an adaptation to the fermentation’s environmental conditions (Rosenstein et al., 2009).
ABC transporter and heme IsdEF (iron-regulated surface determinant) transporter proteins (that were exclusive for pathogenic tested genomes) are required for the mechanism of heme obtaining in S. aureus (Nygaard et al., 2006; Zhu et al., 2008). Iron is a crucial metal for the life-sustaining of pathogenic bacteria and is vital for launching the infection process (Mazmanian et al., 2003; Kuroda et al., 2005). The IsdEF transporter is a surface lipoprotein that binds to heme and works beside the ABC transporter to transport heme into the cytoplasm of bacteria (Zhu et al., 2008). Another comparative study supports our findings as Rosenstein et al. (2010) also found iron uptake systems specific for pathogenic species.
Transposons are mobile genetic elements of bacteria, which our analysis detected as exclusive for pathogenic Staphylococcus. They encode transposase enzymes, act on specific DNA sequences, and insert them into a new target DNA site. Moreover, transposons enhance the genomic diversification of staphylococci species, so the more transposons in the genome, the higher plasticity of the genome is (Baba et al., 2002; Loessner et al., 2002). Non-pathogenic staphylococci are considered relatively more stable due to the lack of transposons in their genomes; such findings emphasize the role of mobile elements in the pathogenicity of Staphylococcus (Rosenstein et al., 2009). The study outcome related to transposons strengthens several previous studies that suggested a role for transposase in spreading the antibiotic resistance gene among different species (Rowland & Dyke, 1989; Ito et al., 2003; Schwendener & Perreten, 2011; Zong, 2013; Harmer & Hall, 2015; Partridge et al., 2018; Guo et al., 2020). Indeed, such features led to the increasing pathogenicity of staphylococci species.
Biosynthetic, cellular biosynthetic, and organic substance biosynthetic process GO terms were significantly under-represented in the pathogenic genomes tested. These terms are associated with the formation of substances required for metabolism. Cellular biosynthetic is involved in creating materials carried out by individual cells. However, the organic substance biosynthetic process is for any molecular entity containing carbon (Binns et al., 2009). As no article researches the biosynthetic process of any of the tested groups nor elaborates on the importance of such a process, the reason behind this underrepresentation still needs to be clarified. Nevertheless, localization, biological regulation, metal ion transport, and DNA recombination are typically expressed in pathogenic Staphylococcus genomes, as reported by several studies (Jin et al., 2014; Liu et al., 2018, 2020) and explains the annotation of these terms to ORFs of pathogenic tested genomes.
The infectious pathway of Staphylococcus is far more complicated and cannot be elucidated in one study. Thus, future studies are recommended with a larger sample size to test the expression level of each group’s uniquely annotated GO terms and deeply investigate their roles.
The importance of our findings emerges from the fact that smORFs have been ignored over the years, although several recent studies have shown their enormous potential (Hobbs, Astarita & Storz, 2010; Khitun, Ness & Slavoff, 2019; Cerqueira & Vasconcelos, 2020). The 23 selORFs features correspond to all smORFs properties. They are small (size <50 amino acids) and highly conservative. Some are nested within genes and predicted to be involved in regulatory function, mostly in the mRNA catabolic process. The mRNA catabolic process occurs in the ribosome during translation elongation and induces pathways of mRNA decay (Hayamizu et al., 2005). Translation stalling occurs when; (i) mRNA is damaged or truncated, (ii) in case of excessive mRNA secondary structure, or (iii) upon which there are insufficient amounts of amino acid or tRNA (Nielsen et al., 2011; Wencker et al., 2021), and stalling regulates the translation of downstream genes (Nakatogawa & Ito, 2002).
Bacteria have a wide range of regulatory mechanisms in their cellular stress response. Several shreds of evidence have proposed that smORFs have a role in cellular stress responses, such as antibiotics, host-infection, and nutrition hemostasis (Kültz, 2005; Hobbs, Astarita & Storz, 2010; Hobbs et al., 2012). The proposed mechanism by which smORFs affect cellular stress response is via both transcriptional and post-transcription regulation pathways. Sigma factor B (SigB) mediates the transcriptional response approach, while sRNA mediates the post-transcriptional regulatory mechanism (Novick, 2003).
SigB contributes to the overall stress response in both staphylococci and bacilli. It regulates several gene transcription expressions, including those encoding virulence factors and biofilm formation in S. aureus (Wu, de Lencastre & Tomasz, 1996). SigB regulates the transcription of alternative frames and intergenic regions (IGRs) in bacteria, leading to harmful control genes in a collateral manner (Wu, de Lencastre & Tomasz, 1996; Bischoff et al., 2004; Miller et al., 2011). In a recent study, researchers identified three sigB-regulated genes within IGRs of S. aureus. Two of these genes contained smORFs encoding putative small proteins, whereas the third transcript was not preceded by a likely ribosomal binding site, suggesting it was a non-coding RNA. However, the transcript overlapped with the mntC gene by approximately 180 nucleotides, indicating a possible cis-acting anti-sense regulatory mechanism (Nielsen et al., 2011). Moreover, an unannotated smORF named gndA was found within the gnd gene. Researchers believed that the gndA is expressed in an alternative reading frame during heat shock in E. coli under the control of the Sig-B factor (Khitun, Ness & Slavoff, 2019).
RNA III belongs to the trans-encoded base pairing small RNAs (sRNAs) (Geisinger et al., 2006). RNA III controls several genes’ expression profiles. It forms an imperfect duplex that targets specific mRNAs and represses their translation (Wadler & Vanderpool, 2007; Nielsen et al., 2011), so far most studied sRNAs in E. coli, B. subtilis, and S. aureus appear to be non-coding.
In line with these elaborations, the selORFs are hypothesised to be transcribed under the regulation of the Sig-B factor in certain conditions, thereby producing non-coding sRNAs, which negatively regulate the transcription of the adjacent gene (Pförtner et al., 2014; Rodriguez Ayala, Bartolini & Grau, 2020). However, this hypothesis first requires the exclusion of false-positive smORFs (Fuchs et al., 2021), then transcriptomics analysis of the verified selORFs in different environments, besides experimentally investigating Sig-B’s role in controlling their transcripts.
Genome assembly database - RefSeq accessions: Staphylococcus aureus subsp. aureus Mu3 genome assembly ASM1044v1. https://identifiers.org/refseq.gcf:GCF_000010445.1
Genome assembly database - RefSeq accessions: Staphylococcus lugdunensis HKU09-01 genome assembly ASM2508v1 https://identifiers.org/refseq.gcf:GCF_000025085.1
Genome assembly database - RefSeq accessions: Staphylococcus haemolyticus JCSC1435 genome assembly ASM986v1. https://identifiers.org/refseq.gcf:GCF_000009865.1
Genome assembly database - RefSeq accessions: Staphylococcus saprophyticus subsp. saprophyticus ATCC 15305 = NCTC 7292 genome assembly ASM1012v1. https://identifiers.org/refseq.gcf:GCF_000010125.1
Genome assembly database - RefSeq accessions: Staphylococcus schleiferi genome assembly ASM118885v1 https://identifiers.org/refseq.gcf:GCF_001188855.1
Genome assembly database - RefSeq accessions: Staphylococcus carnosus subsp. carnosus TM300 genome assembly ASM940v1. https://identifiers.org/refseq.gcf:GCF_000009405.1
Genome assembly database - RefSeq accessions: Staphylococcus cohnii. Genome assembly ASM199020v1. https://identifiers.org/refseq.gcf:GCF_001990205.1
Genome assembly database - RefSeq accessions: Staphylococcus warneri SG1 genome assembly ASM33273v1. https://identifiers.org/refseq.gcf:GCF_000332735.1
Genome assembly database - RefSeq accessions: Staphylococcus nepalensis genome assembly ASM290274v1. https://identifiers.org/refseq.gcf:GCF_002902745.1
Genome assembly database - RefSeq accessions: Staphylococcus pasteuri genome assembly ASM244291v1. https://identifiers.org/refseq.gcf:GCF_002442915.1
NCBI Reference Sequence: Salinicoccus alkaliphilus DSM 16010, whole genome shotgun sequence. Accession number NZ_FRCF01000009.1; https://identifiers.org/refseq:NZ_FRCF01000009.1
NCBI Reference Sequence: Salinicoccus albus DSM 19776 strain YIM-Y21 G343DRAFT_scaffold00006.6_C, whole genome shotgun sequence. Accession number NZ_ARQJ01000028.1; https://identifiers.org/refseq:NZ_ARQJ01000028.1
NCBI Reference Sequence: Salinicoccus carnicancri Crm 50.SCCRM.1_10, whole genome shotgun sequence. Accession number NZ_ANAM01000010.1; https://identifiers.org/refseq:NZ_ANAM01000010.1
NCBI Reference Sequence: Nosocomiicoccus ampullae strain DSM 19163 Ga0415238_04, whole genome shotgun sequence. Accession number NZ_JACHHF010000004.1; https://www.ncbi.nlm.nih.gov/nuccore/NZ_JACHHF010000004.1
NCBI Reference Sequence: Nosocomiicoccus massiliensis isolate MGYG-HGUT-01449, whole genome shotgun sequence. Accession number NZ_CABKSY010000018.1; https://www.ncbi.nlm.nih.gov/nuccore/NZ_CABKSY010000018.1
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)