Keywords
Malaria treatment, drug design, molecular docking, molecular quantum similarity, chemical reactivity indices, Density Functional Theory.
This article is included in the Cheminformatics gateway.
Malaria treatment, drug design, molecular docking, molecular quantum similarity, chemical reactivity indices, Density Functional Theory.
Malaria remains a substantial global health challenge, particularly in numerous tropical and subtropical regions. The disease is endemic in various countries, especially in sub-Saharan Africa, Southeast Asia, and parts of Central and South America,1–3 where it imposes the highest disease burden. Plasmodium parasites, transmitted through the bites of infected female Anopheles mosquitoes, cause malaria. Among the Plasmodium species infecting humans, P. falciparum is the most lethal. In 2020, the global tally of malaria cases, as reported by the World Health Organization (WHO), stands at approximately 241 million, with 627,000 recorded deaths, with a disproportionate impact on Africa.3–6 At-risk demographics, including infants and expectant mothers, bear a significant burden, with pregnant women facing heightened risks, including maternal anemia and low birth weight. Control and prevention measures encompass the use of insecticide-treated bed nets, indoor residual spraying, antimalarial drugs, and initiatives targeting mosquito breeding sites.6,7 While Artemisinin-based combination therapies (ACTs) are extensively employed for treating uncomplicated malaria, other antimalarial drugs may be utilized for specific cases or regions. The emergence of drug resistance, particularly in the Greater Mekong Subregion, presents a formidable challenge to malaria control efforts.7–10 Additionally, concerns about insecticide resistance in mosquitoes underscore the importance of exploring new alternatives for malaria treatment.11–14
Current research is dedicated to advancing the development of novel antimalarial medications, vaccines, and inventive vector control strategies. The objective is to enhance the arsenal of tools for preventing, treating, and ultimately eradicating malaria. Global malaria control initiatives are actively supported by international organizations, governments, and non-governmental organizations, with pivotal roles played by the WHO's Global Malaria Program and the Roll Back Malaria Partnership in orchestrating collaborative endeavors to diminish the worldwide impact of malaria.15–18
Furthermore, the challenges posed by the COVID-19 pandemic have impacted malaria control efforts, redirecting resources and straining health systems. For the most recent and accurate information, it is advisable to refer to recent reports from health organizations and institutions actively engaged in malaria control.19–22 Protein kinases play pivotal roles in diverse cellular processes, encompassing signal transduction, cell cycle regulation, and gene expression.23,24 Within the context of the malaria parasite, Plasmodium spp., protein kinases are indispensable for the parasite's survival and proliferation within the host.25–29 Understanding the role of protein kinases in the malaria parasite holds significance for the development of targeted therapeutic strategies. Here are key insights into protein kinases in the malaria parasite:
Abundance of Protein Kinases: The genome of Plasmodium spp., including the human malaria parasite Plasmodium falciparum, encodes a multitude of protein kinases. P. falciparum, exemplarily, boasts over 80 putative protein kinases.30–33 Diverse Functions: Protein kinases in Plasmodium participate in a spectrum of cellular processes, including signal transduction, cell cycle progression, host cell invasion, and immune evasion.33,34 They play a crucial role in regulating the parasite's life cycle, spanning the invasion of host cells by the sporozoite stage to the replication within hepatocytes (liver stage) and erythrocytes (blood stage).35–37 Unique Kinases: Some protein kinases in the malaria parasite exhibit uniqueness or distinct features compared to their counterparts in the host.38–40 This distinctiveness presents opportunities for developing drugs that selectively target the parasite without affecting host cell kinases.40–42
Druggability and Therapeutic Targets: Protein kinases have been identified as potential drug targets for antimalarial therapies. Inhibiting specific kinases can disrupt essential cellular processes in the parasite, leading to its demise. Ongoing efforts aim to identify and characterize specific protein kinases critical for the parasite's survival and development.43 Inhibitors targeting these kinases could serve as the foundation for developing new antimalarial drugs. Examples of Protein Kinases in Plasmodium: Notable examples include PfCDPK1 (Calcium-Dependent Protein Kinase 1), involved in the invasion of host erythrocytes; PfPKA (Protein Kinase A), with a role in the regulation of the cell cycle; and PfPKG (Protein Kinase G), significant for the development of sexual stages of the parasite in the mosquito vector.40–43
Progress in antimalarial drug discovery has been notable, but challenges such as drug resistance and the intricate life cycle of the malaria parasite persist, driving ongoing research efforts.38–41 Key aspects and approaches in antimalarial drug discovery include:
Targeting the Parasite's Life Cycle: Malaria, caused by Plasmodium parasites, involves a complex life cycle in both the mosquito vector and human host. Antimalarial drug discovery aims to target various stages, including the liver stage, blood stage, and transmission-blocking interventions to disrupt the life cycle.30–35 Artemisinin-Based Combination Therapies (ACTs): Artemisinin and its derivatives are potent antimalarial drugs that rapidly reduce parasite biomass. ACTs, combining artemisinin compounds with other antimalarials, have proven highly effective in treating uncomplicated malaria.39–42 However, concerns arise over the emergence of artemisinin resistance in some regions. Drug Resistance Monitoring and Surveillance: The development of resistance to antimalarial drugs is a significant challenge. Ongoing efforts focus on monitoring and surveillance, utilizing molecular techniques to identify resistance markers in the parasite's genome.40–43 New Target Identification: Researchers continually identify potential drug targets within the malaria parasite. This includes enzymes in essential metabolic pathways, proteins crucial for invasion and egress from host cells, and unique features of the parasite's biology exploitable for drug development.25–30 High-Throughput Screening (HTS): HTS techniques rapidly test large compound libraries to identify molecules with antimalarial activity, expediting the drug discovery process.30–35 Fragment-Based Drug Design: This methodology involves screening small molecular fragments for their ability to bind to a target, leading to the design of more potent and specific compounds.35–38 Repurposing Existing Drugs: Cost-effective repurposing of drugs developed for other diseases has shown promise. Some drugs, initially designed for different purposes, exhibit antimalarial activity, expediting the development process. Vaccines and Transmission-Blocking Strategies: Malaria vaccine development, while not conventional, is integral to the broader strategy against the disease.40–43 Additionally, transmission-blocking drugs aim to reduce mosquito transmission of the parasite, contributing to malaria control efforts. Public-Private Partnerships: Collaboration among academic institutions, pharmaceutical companies, and public health organizations is crucial for advancing antimalarial drug discovery. Public-private partnerships facilitate pooling resources, expertise, and funding to accelerate research and development.38–43
In this manuscript is used an approach based in molecular docking, Molecular quantum similarity and Chemical reactivity indices. A comprehensive approach could involve integrating molecular docking, molecular quantum similarity, and chemical reactivity indices. For example, molecular docking can be used to predict the binding mode of a ligand to a target protein, while molecular quantum similarity and reactivity indices can help understand the electronic and chemical properties governing the interaction. We Started with molecular docking to identify potential ligands, validating the results using quantum similarity methods, and then analyze the reactivity indices to gain insights into the chemical behavior of the molecules.
Integrating these approaches allows us to leverage the strengths of each method, providing a more holistic understanding of molecular interactions and properties. However, it's important to note that these computational techniques are complemented by experimental validation for robust results. In this sense, the ligand studied has been analyzed by the Zhang group.44 In this sense, in this manuscript these ligands will be analyzed using new reported protein kinases with the aim of obtaining new considerations on the antimalarial activity of these ligands.
During the docking experiment, the receptor structure was generated in adherence to specific protocols, drawing inspiration from the crystal structure of the co-crystallized ligand located at the core of PDB entries 1ob3 and 2pmn. Structural adjustments were implemented employing the protein preparation wizard module from the Schrödinger suite 2017-1. These adjustments included optimizing the hydrogen bond (H-bond) network and refining the protein structure. The PropKa (https://github.com/schrodinger/propka-3.1) utility was utilized to determine protonation states at physiological pH. Additionally, restrained molecular minimization was executed through the Impact Refinement (Impref, https://newsite.schrodinger.com/platform/products/maestro/) module, with heavy atoms constrained to a low root-mean-square deviation (RMSD, https://newsite.schrodinger.com/platform/products/maestro/) from the initial coordinates.45–47
In contrast, the molecular structures of the compounds were fashioned using the Maestro Editor (Maestro, version 11.1, Schrödinger, LLC). Subsequently, 3D conformations were generated employing the LigPrep module, and ionization/tautomeric states were predicted under physiological pH conditions using Epik. Finally, energy minimization was carried out using the Macro model and the OPLS2005 force field.
For the molecular docking investigations, Glide48,49 was employed with default parameters and the Standard Precision (SP) model. The docking grid was established using default settings, positioning the co-crystallized ligands from PDB entries 1ob3 and 2pmn at the center. To enhance the binding of larger ligands, a scaling factor of 0.8 for the van der Waals radii of nonpolar protein atoms was applied. Extra precision (XP) was also employed to accommodate alternate receptor conformations suitable for binding ligands with unconventional orientations through induced fit docking (IFD). This approach allows for potential side-chain, backbone, or combined movements in the protein structure upon ligand docking. Redocking was performed for all results, and subsequent RMSD calculations were conducted. The binding pocket was identified using Glide.48,49
The docking procedure comprised four meticulous steps, leveraging Glide's scoring function and Prime's advanced conformational refinement for precision:
i. Initial docking was conducted using Glide on the rigid receptor to generate a set of poses.
ii. The side-chain prediction module of the Prime tool was utilized to sample the protein, followed by structural minimization for each pose of the protein/ligand complex.
iii. Redocking of the ligand into the low-energy induced-fit structures from the previous step was carried out using Glide's default parameters, without van der Waals scaling.
iv. The binding energy (IFDScore) was estimated, taking into account the docking energy (GScore), receptor strain, and solvation terms (Prime energy).
To further evaluate ligand interactions in the active site, the extent of residue movement induced by the induced-fit docking (IFD) computation was considered. For both the most and least active ligands, all poses were compared within the molecular set. Molecular dynamics calculations spanning 30 ns were employed to predict the best poses and analyze their stabilization in the active site.
The Molecular Quantum Similarity Measure (MQSM), represented as ZAB, is utilized to compare two systems, A and B, by evaluating molecules generated using their respective Density Functions (DFs). The DFs are multiplied and integrated over electronic coordinates, incorporating weighting by a pre-established positive operator Ω(r1, r2)50–52:
The selection of the operator in Equation 1 holds significance, as it shapes the information under comparison and functions as a similarity measure between the two systems. For example, the Dirac delta function (Ω(r1, r2) = δ(r1 - r2)) proves effective for functions characterized by high peak values, such as electronic density, resembling a charge or point mass.
Commonly utilized similarity metrics include the overlapping Molecular Quantum Similarity Measure (MQSM) and the Coulomb operator (Ω(r1, r2) = |r1 - r2|−1). Additionally, a self-similarity measure (ZAA for molecule A) can be applied, even when comparing two equivalent molecules.52
For a group of N molecules, a similarity measure is computed for each molecule concerning others, resulting in a symmetric matrix. Each column in this matrix represents the similarity measurements between the molecule and every constituent in the group, generating discrete N-dimensional representations for each structure. These vectors serve as unique chemical descriptors extending beyond traditional molecular descriptors.51–58 They are universal, derived from any collection of molecules. They are impartial, determined solely by density functions and similarity measurements.
Carbó's similarity index (Equation 2) for two molecules, I and J, relies on the cosine of the angle formed by their density functions when treated as vectors. This index varies between 0 and 1, signifying the degree of similarity between the two molecules.50–55
Carbó's similarity index between two molecules, I and J, is obtained from Equation 2. Also known as the cosine similarity index, it aligns with the cosine of the angle formed by the density functions treated as vectors. The Carbo QSI for any pair of compared molecules spans from 0 to 1, indicating the extent of similarity between the two molecules (approaching 1 when I equals J).50–55
Equation 3 introduces a similarity measure expressed as the Euclidean distance index (Equation 4). This index represents the distance or dissimilarity between two quantum objects, allowing geometric interpretation based on the norm of the differences in density functions:
It is simplified to the so-called Euclidean distance index when k = x = 2. Index 3 of the form can also be defined as follows:
This Equation 4 forms the distance index of infinite order.50–55
The Dirac delta distribution, Ω (r1, r2) = δ (r1, r2), stands out as the most common and intuitive option for a positively defined operator in this context. Such choices refine the broad definition of MQSM, resulting in the computation of the overlap MQSM, which gauges the volume measurements covered by both electronic density functions when superimposed.56–60
Derived from its physical definition, the Dirac delta function is computationally manageable. The Molecular Quantum Similarity Measure (MQSM) gauges the degree of overlap in molecular comparisons by incorporating information about the electron concentration within the molecule.61–63
When the operator (Ω) is replaced with the Coulomb operator, Ω (r1, r2) = , the coulomb MQS is generated, which defines the electrostatically repellent coulomb energy between two charge densities64–67:
The Coulomb operator plays a role in influencing the overlap of density functions. When molecular density functions are considered as electron distributions in space, this equation essentially extends the Coulomb operator for continuous charge distribution. In certain cases, it can be used as electrostatic potential descriptors. This operator is associated with electrostatic interactions and measures the electrostatic repulsion between electronic distributions.68
Another major transformation that can be expressed in terms of the classical distance is:
Here is the distance between a and b, and k = 2 is the definition of distance, respectively. Subsequently, the Euclidean distance between A and B as two quantum objects are defined by68–70:
Occasionally it is written as: , where DAB has values in the range of [0,∞) but for earlier circumstances where the compared items are identical, it converges to zero between them70–72:
The norm of the differences in the density functions of the compared objects can be used to interpret this index geometrically. The distance or dissimilarity index can be used to define the Euclidean distance index, which can also be represented as73:
In this investigation, the TGSA is employed for data alignment, aligning molecules based on a common skeleton using atomic types and interatomic bonding interactions. TGSA functions through the scrutiny of atomic pairs, forming triangles, and evaluating atomic triads. This approach offers a distinctive and chemically intuitive alignment, even in cases where molecular structures exhibit limited flexibility.73–76
Research in this domain has unveiled a firmly established correlation between quantum similarity and descriptors associated with chemical reactivity.77,78 Both quantum similarity and Density Functional Theory (DFT) make use of the density function as a fundamental component in the analysis of similarity indices. Specifically, the Coulomb index can be correlated with electronic factors influencing chemical reactivity. In the computation of global reactivity indices, such as chemical potential (μ),77 hardness (ɳ),78 and electrophilicity (ω),79,80 Frontier Molecular Orbitals (FMO) and the energy gap will be employed. These indices, represented by Equations 11-13, offer valuable insights into system stability. Chemical potential gauges the tendency of electrons to deviate from the equilibrium system,81 while chemical hardness assesses a chemical species' resistance to altering its electronic configuration.
The mathematical definition of the electrophilicity index (ω) is related to the stabilization energy of a system when it becomes saturated by electrons from the external environment.39,40:
In this study, the focal descriptors of reactivity under examination were the Fukui functions. Equation (14) depict how the electronic density of the system responds to changes in the global charge, representing the derivative of the electronic density with respect to the electron count under a uniform external field.
The terms and and are utilized to represent nucleophilic and electrophilic attacks, respectively.81–83
This approach integrates both global and local reactivity descriptors to evaluate quantum similarity within the molecular set. All calculations were conducted using the B3LYP method84 with the 6-311xxG(d,p) basis set,84 which represents an improvement over the 6-311G(d) basis set. This enhancement facilitates the computation of electronegativity, hardness, reactivity indices, and frontier molecular orbitals at a level of quality comparable to larger basis sets like Aug-cc-pVQZ and Aug-cc-pV5Z. The Gaussian 16 package85 was employed in conjunction with this method/basis set combination.
Molecular docking plays a crucial role in drug discovery for malaria treatment. Malaria, caused by the Plasmodium parasites, A potentially fatal illness that impacts millions of individuals globally. Developing effective drugs to combat malaria requires a deep understanding of the interactions between potential drug molecules and target proteins within the Plasmodium parasites.
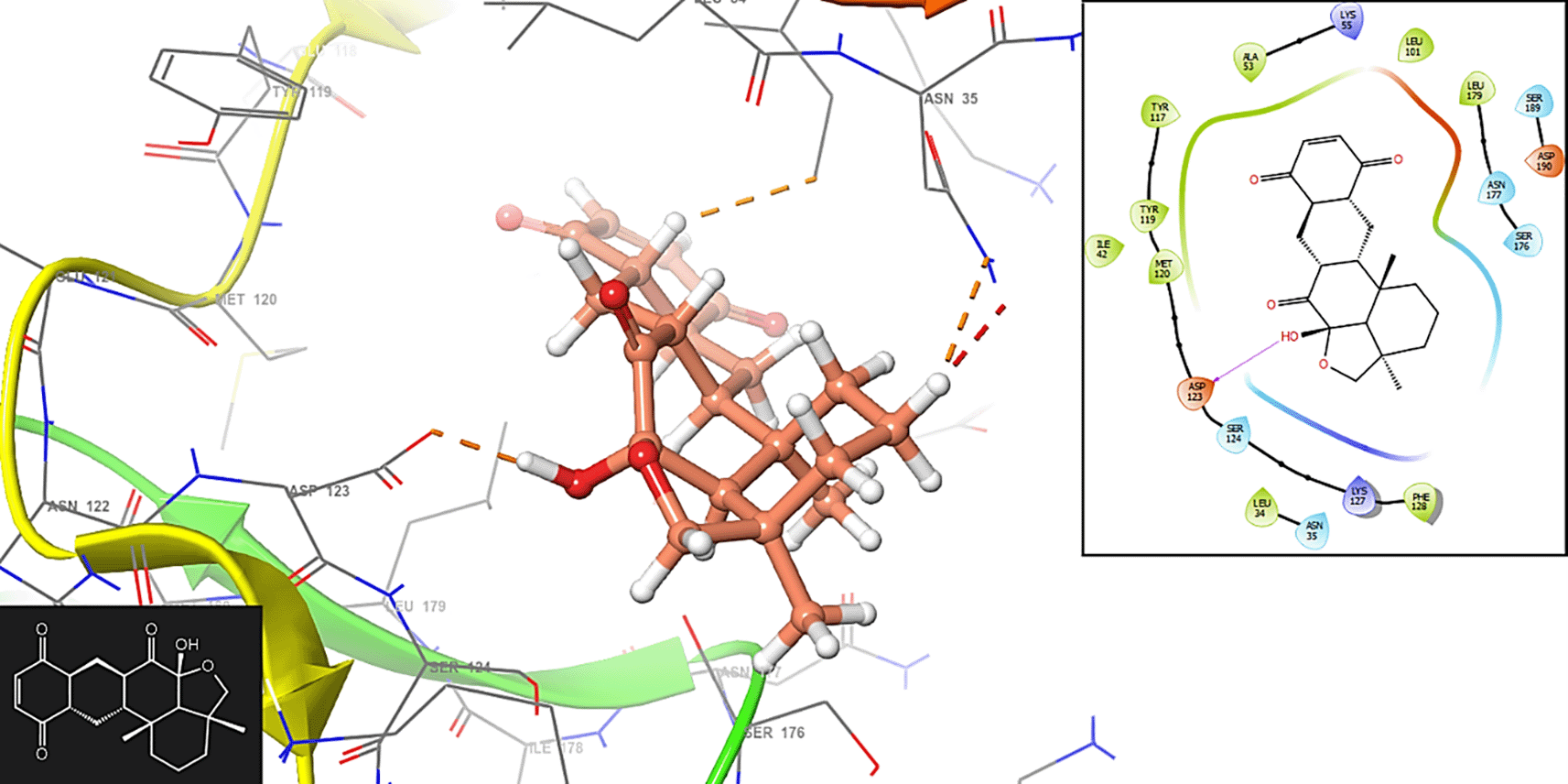
The compound 1 has interaction with the residue MET120 (-H, 1,125 Å) (Figure 1). In Figure 2 is shows the interaction with the compound 2 with the residues ASP123 (-H, 1,148 Å) and SER (-H, 1,358 Å).
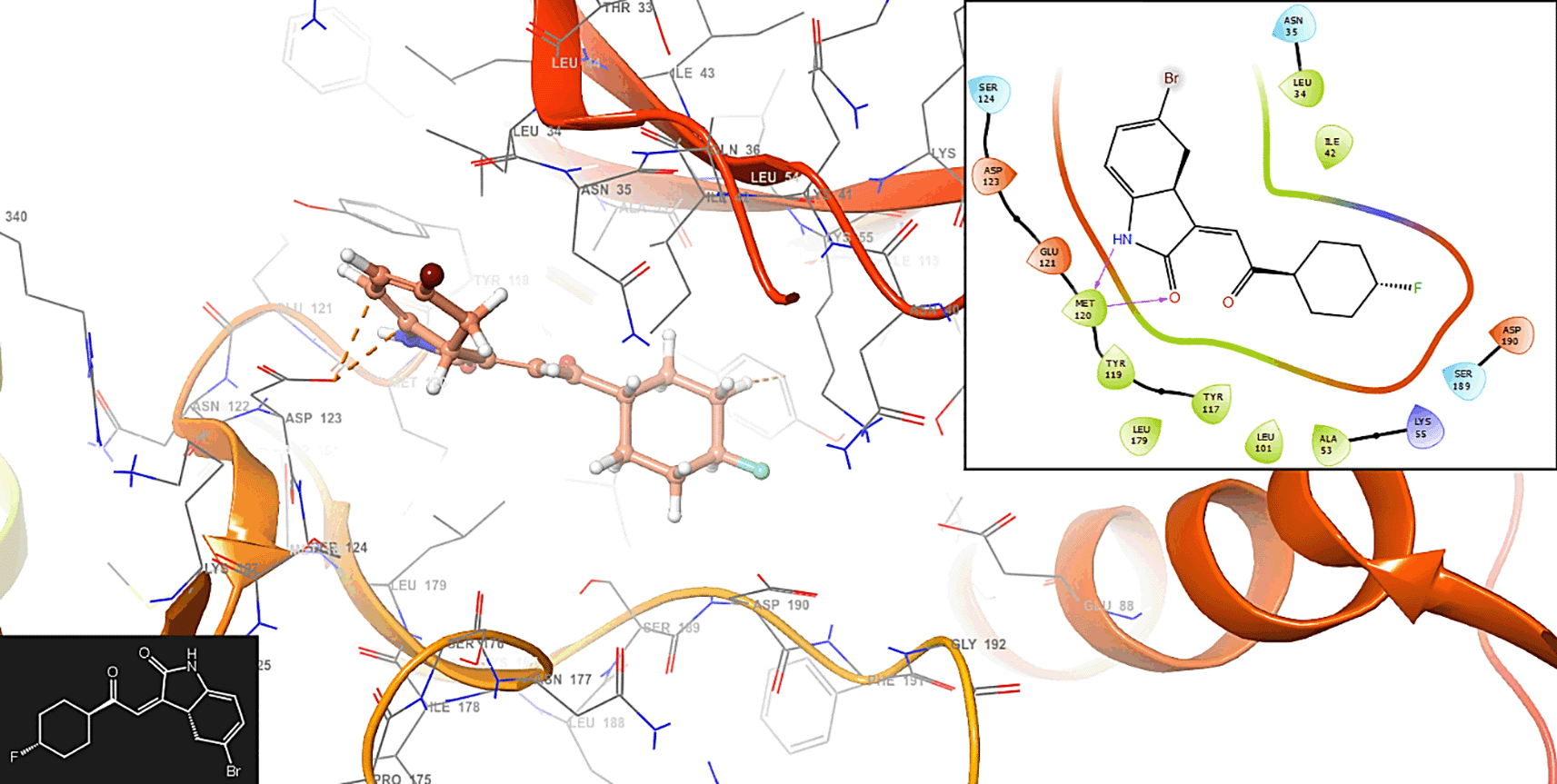
In Figure 3, the compound 3 has interaction with the residue ASP123 (-H, 1,2561 Å).
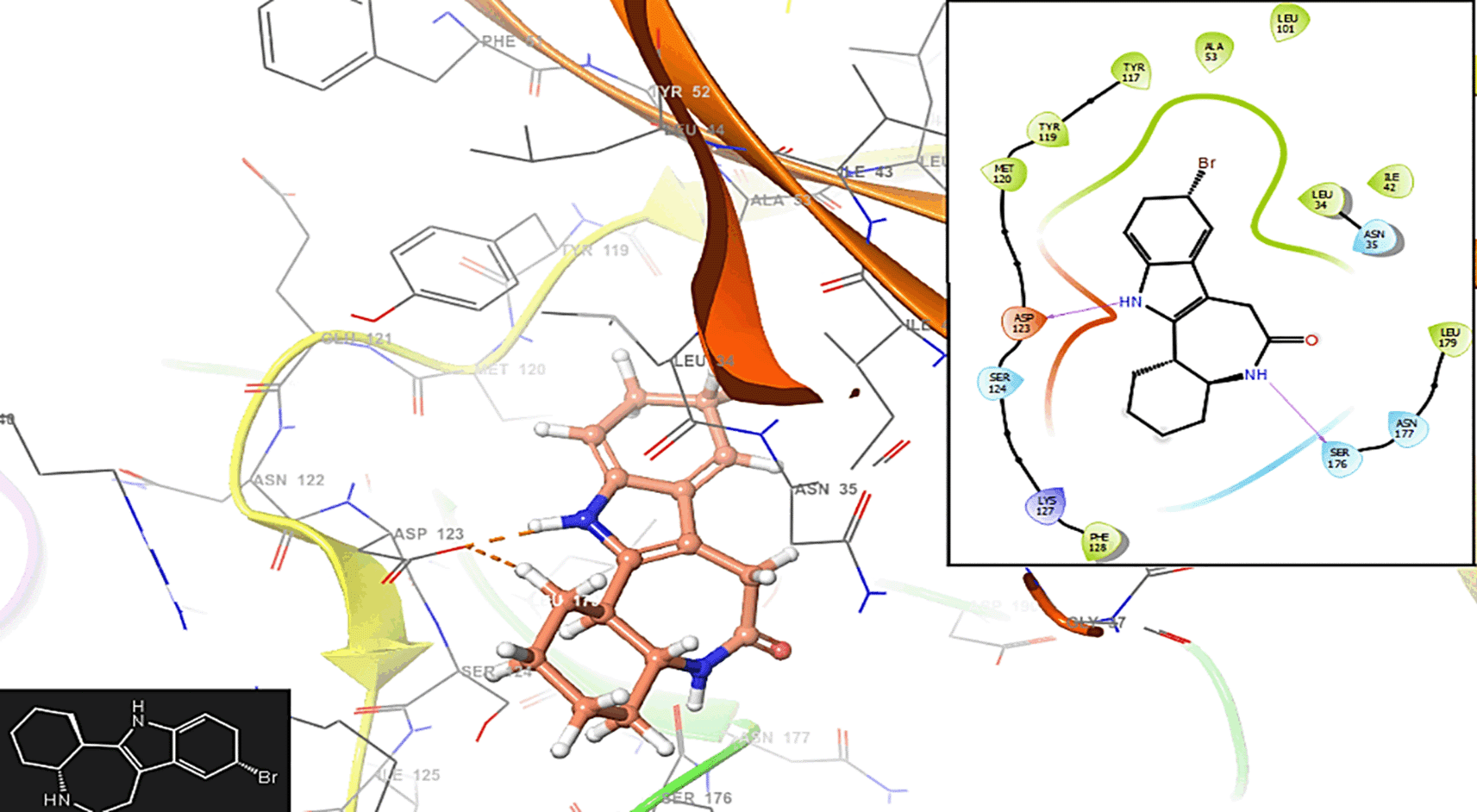
Molecular docking helps identify and validate potential drug targets within the Plasmodium parasites. By simulating the interaction between small molecules and target proteins, researchers can prioritize which proteins are most promising for drug development. The three-dimensional structures of target proteins involved in the malaria-causing parasites are essential for designing drugs that specifically interact with these proteins. Molecular docking aids in the rational design of drug candidates by predicting the binding modes and affinities of small molecules to target proteins. Figure 4 has the interactions with the residues MET120 (-H, 1,1961 Å).
In Figure 5 is shows the interaction with the residue SER176 (-H, 1,256 Å).
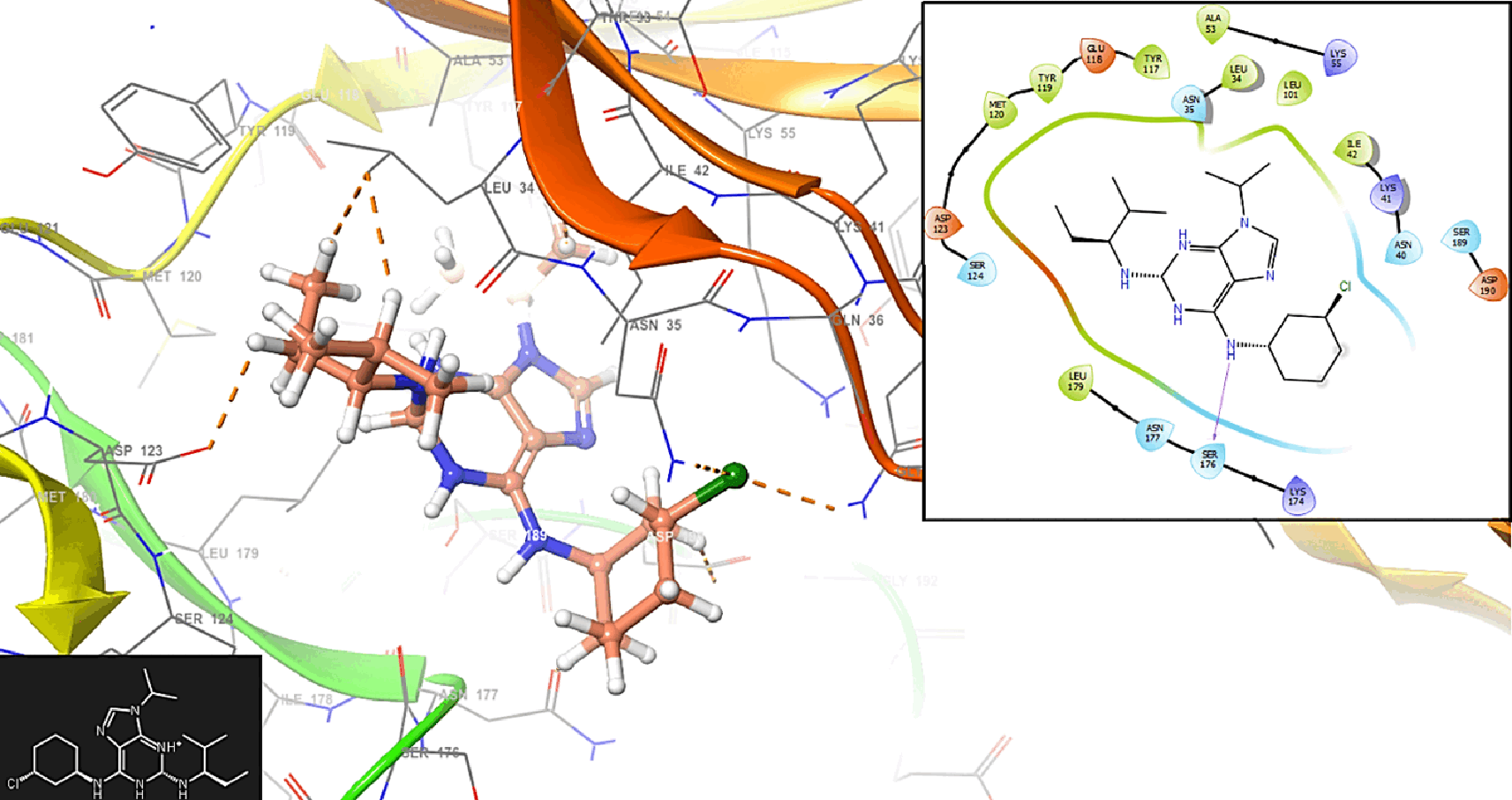
The importance of stabilization on the active site of proteins involved in malaria lies in the context of drug discovery and the development of therapeutics. The active site of a protein is a region where interactions with other molecules, such as substrates or ligands, occur. In the case of proteins associated with malaria, targeting the active site is crucial for designing drugs that can disrupt or modulate the function of these proteins. The active site of a protein is often the functional region where the protein interacts with its substrates or other molecules. By stabilizing the active site, researchers can design drugs that specifically bind to and modulate the activity of the protein involved in the malaria-causing parasite. Stabilizing the active site allows for the design of drugs that are specific to the target protein. This specificity is crucial to avoid unintended interactions with other proteins in the host organism, minimizing side effects and improving the selectivity of the drug.
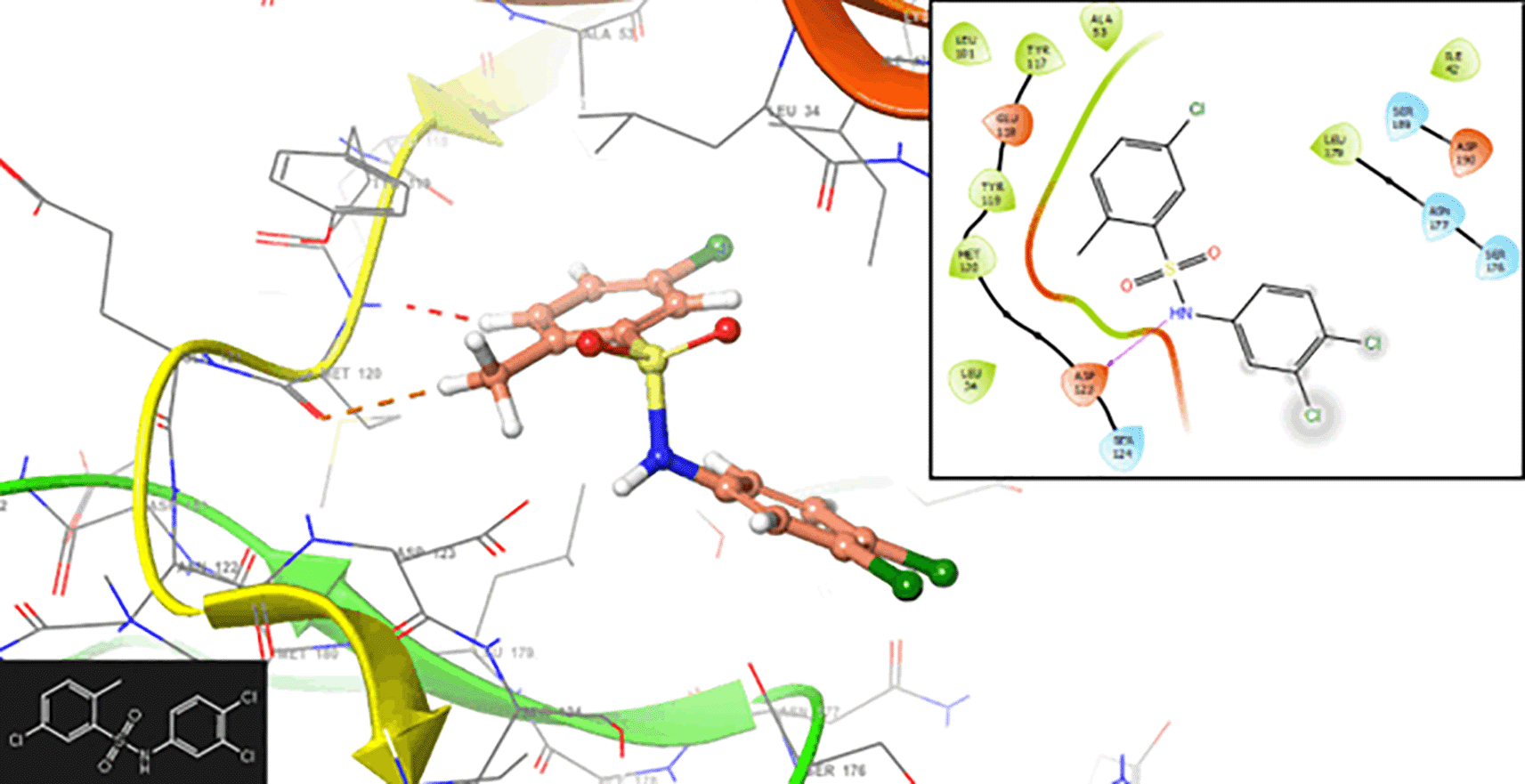
Many drugs work by inhibiting the function of a target protein. Stabilizing the active site facilitates the design of molecules that can bind tightly to the protein, disrupting its normal function. This inhibition is essential for interfering with the life cycle of the malaria parasite or preventing its ability to infect and survive within the host. Some proteins involved in malaria may rely on interactions with other proteins for their function. Stabilizing the active site can disrupt these protein-protein interactions, leading to the inhibition of essential pathways or processes required for the survival of the parasite. Figure 6 shows the interaction with the residues ASP123 (-H, 1,356 Å), SER124 (-H, 1,568 Å) and SER176 (-H, 1,239 Å), respectively.
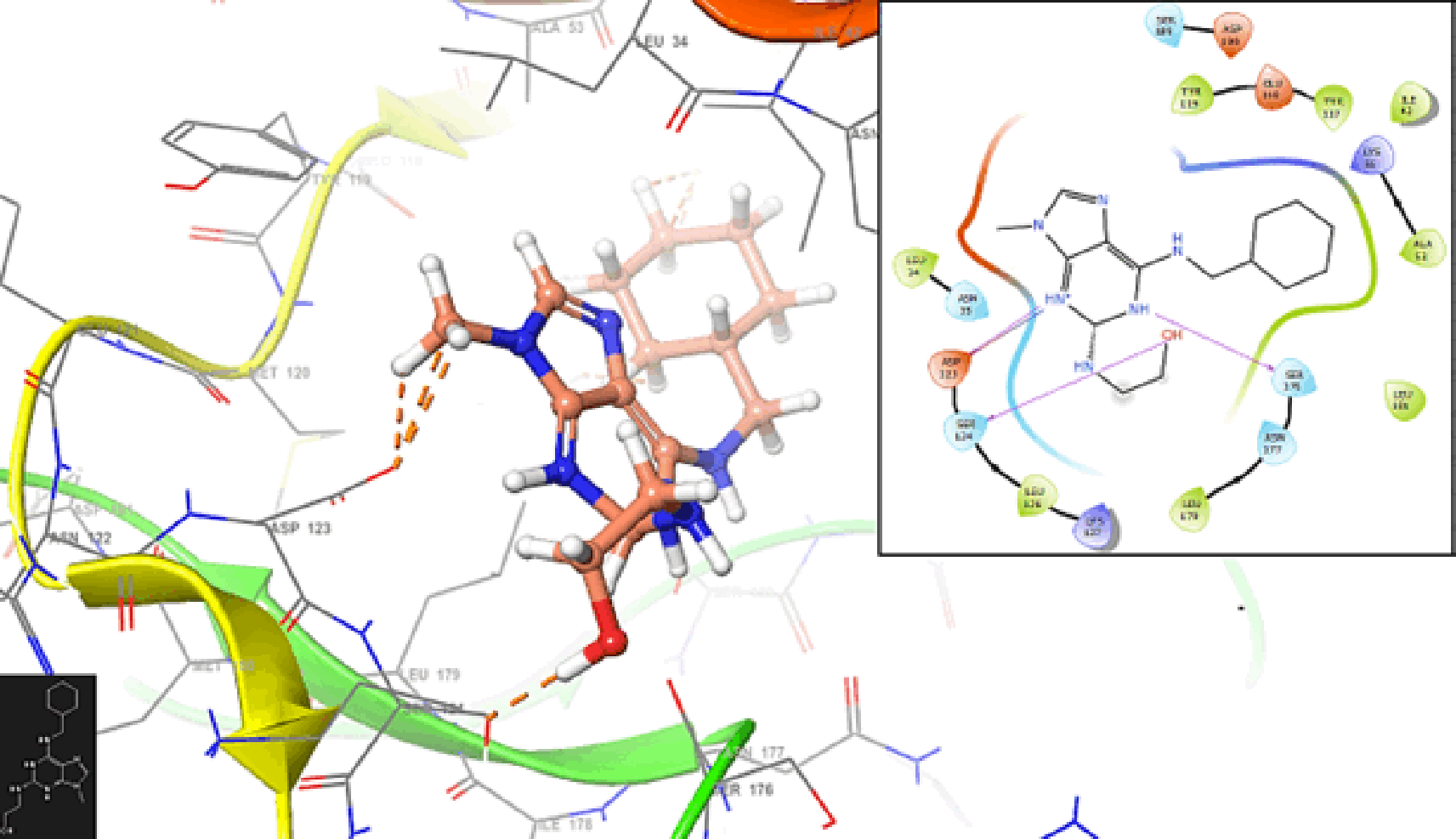
Molecular docking provides insights into the strength of the interactions between drug candidates and target proteins by predicting binding affinities. This information is crucial for selecting lead compounds with high binding affinity and therapeutic efficacy. On the other hand, Malaria parasites can develop resistance to drugs over time. Molecular docking helps researchers understand the structural basis of drug resistance by predicting how mutations in target proteins might affect drug binding. This information is vital for designing drugs that are less prone to resistance.
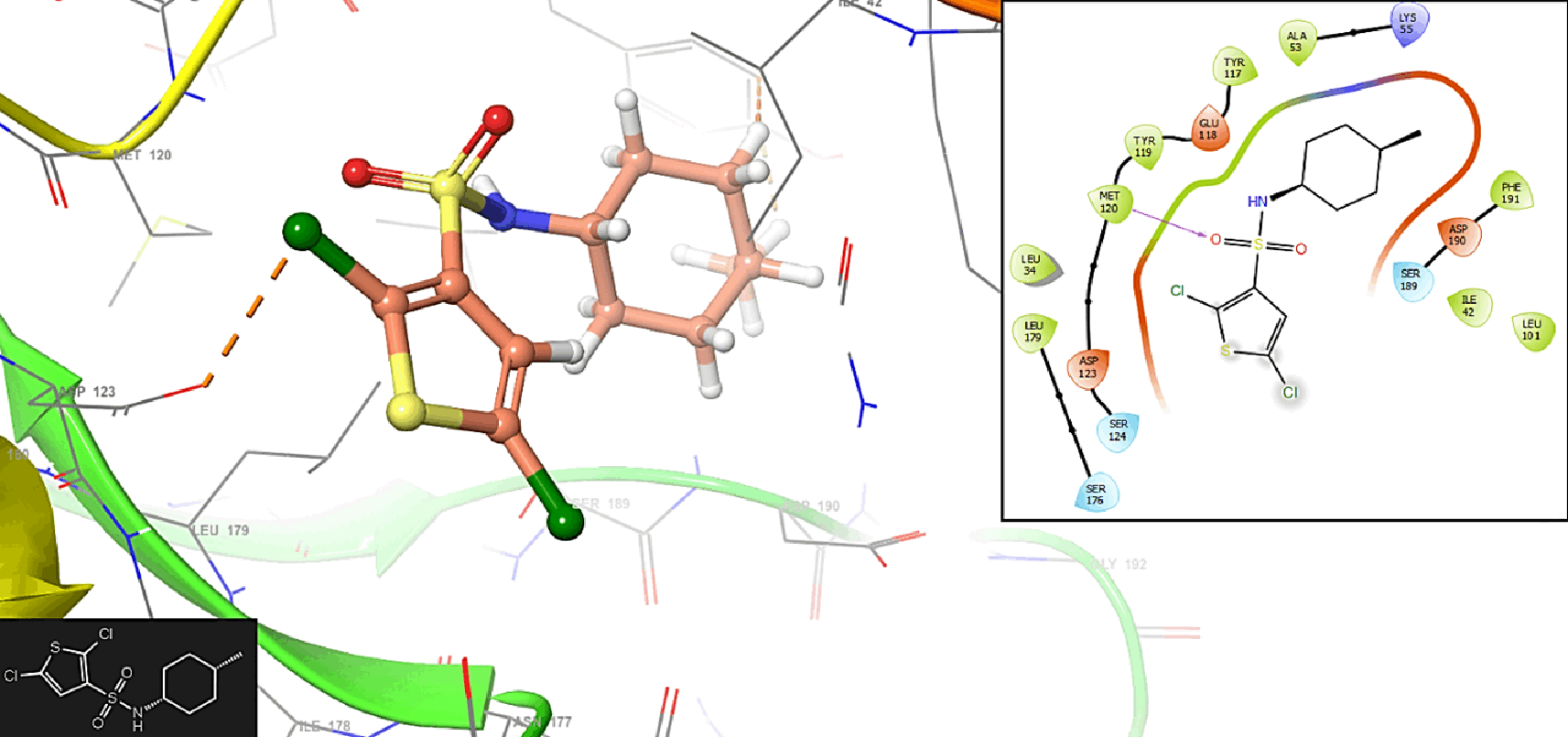
Figure 7 shows the interaction with the residue ASP123 (-H, 1,569 Å) and PHE128 (-H, 1,4265 Å). In Figure 8, we can see the interactions with the residues ASP123 (-H, 1,4569 Å) and PHE128 (-H, 1,3569 Å).
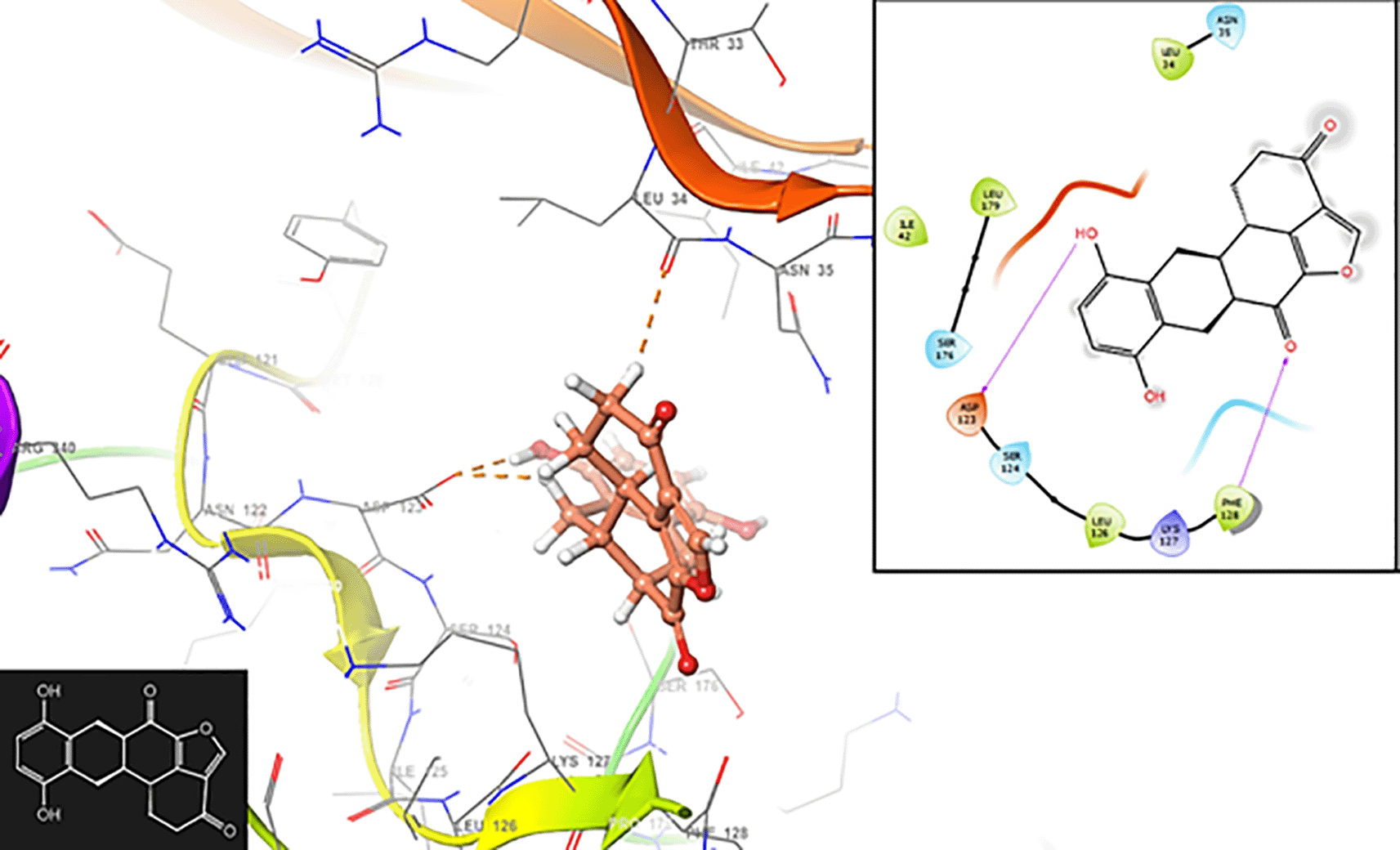
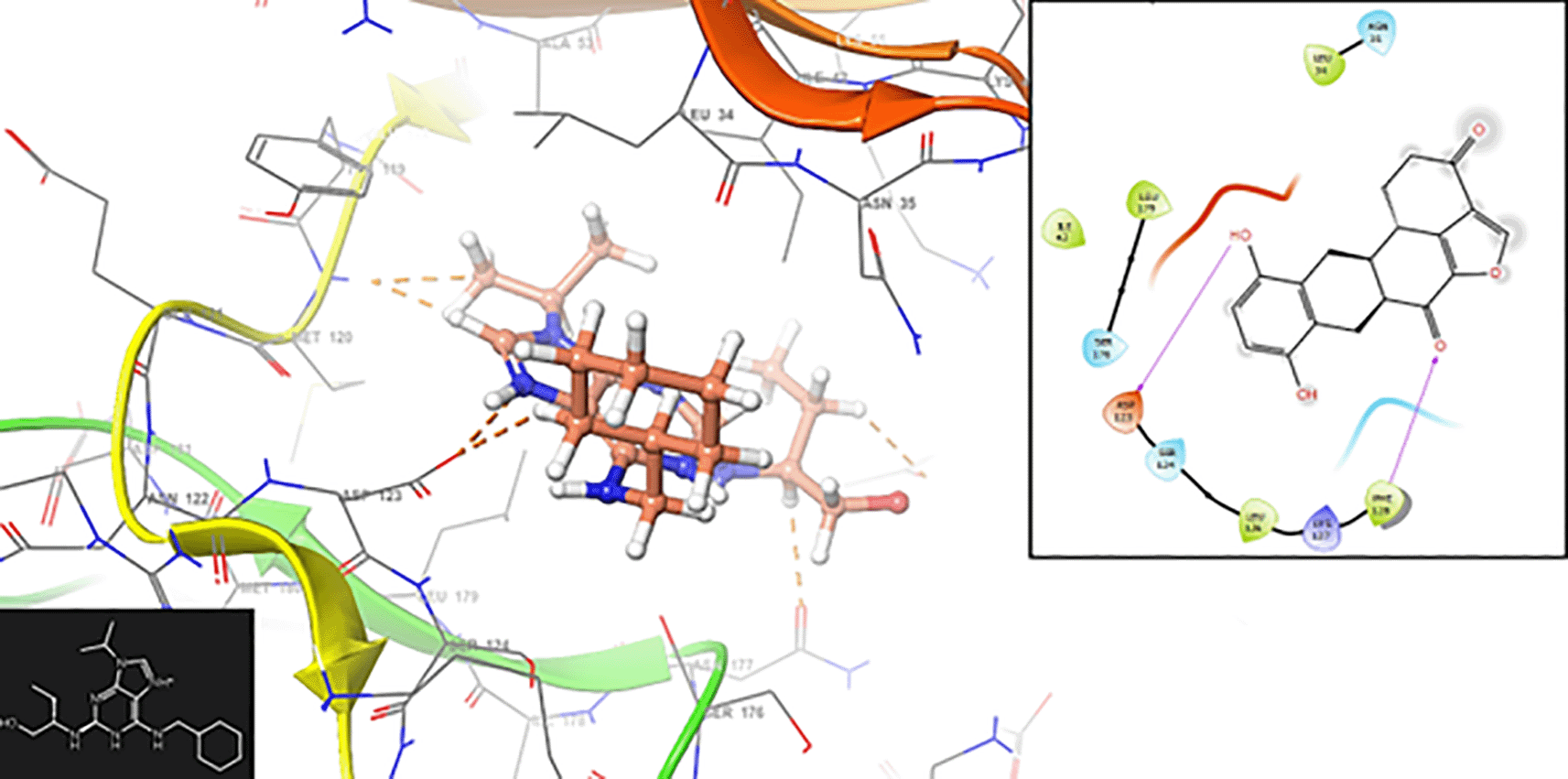
In Figure 9, we can see the interaction with the residue ASP123 (-H, 1,319 Å).
Stabilizing the active site contributes to enhancing the binding affinity between the drug and the target protein. A strong and stable interaction is crucial for the drug to exert its therapeutic effects at lower concentrations, improving the efficacy of the drug. Malaria parasites can develop resistance to drugs over time. By targeting the active site and stabilizing interactions, researchers can design drugs that are less prone to resistance, as mutations in the active site that confer resistance may have a higher fitness cost to the parasite.
Knowledge of the three-dimensional structure of the active site is essential for structure-based drug design. Stabilizing interactions within the active site allows for the rational design of drug candidates that fit precisely into the binding pocket of the target protein. In these sense, stabilizing the active site of proteins involved in malaria is crucial for designing effective and specific drugs. This approach allows for the development of therapeutics that selectively target the parasite, disrupt essential processes, and minimize the risk of resistance. Structure-based drug design techniques, including molecular docking and computational simulations, often play a significant role in understanding and optimizing the stabilization of the active site.
Quantum Similarity Analysis in drug design provides a powerful tool for understanding molecular similarity at a deep, electronic level, contributing valuable insights into the development of new pharmaceutical agents. It integrates quantum mechanical principles into the drug discovery process, offering a more nuanced perspective on molecular interactions and reactivity.
In Table 1 the Higher similarity using the Overlap operator is between the compounds 3 and 4 with 0,8576 and an euclidean distance of 2,0743 (see Table 2). The lowest similarity value is between the compounds 3 and 6 with 0,2666 and an euclidean distance of 4,9818.
In Table 3. The higher similarity using the Coulomb operator is between the compounds 5 and 6 with 0,9389 and an euclidean distance of 21,8492 (see Table 4). On the other hand, the lowest similarity value is between the compounds 4 and 8 with 0,5758 and an euclidean distance of 43,5506.
Because the highest values of electronic quantum similarity were obtained, we have performed an analysis of chemical reactivity to obtain new electronic considerations in the molecular set.
Local reactivity indices guide the introduction of specific functional groups at strategic positions to enhance or modify the biological activity of a molecule. In these sense, global and local chemical reactivity indices play a crucial role in drug design by providing valuable information about the electronic structure and reactivity of molecules. These indices guide the selection of molecular modifications to optimize the pharmacological properties of drug candidates.
In Table 5, the compound with higher chemical potential is the compound 5 with μ = -5,4916 eV, Hardness, (ƞ = 4,5876 eV), Softness, (S = 0,2180 eV)−1 and electrophilicity, (ω = 3,6480 eV). Another compound with good stability is the compound 3 with μ = -4,4106 eV, Hardness, (ƞ = 5,3315 eV), Softness, (S = 0,1876 eV−1) and Electrophilicity, (ω = 1,8243 eV).
The compound with higher electrophilicity is 4 with ω = 4,5777eV. Understanding electrophilicity at the active site of a protein is essential in drug design and medicinal chemistry. The active site is a region within a protein where specific interactions occur with ligands, substrates, or inhibitors. Electrophilicity in this context refers to the propensity of a molecule or a functional group to accept electrons, and it plays a crucial role in the binding of ligands to the protein. Certain amino acid residues in the active site of a protein may exhibit electrophilic characteristics. Common electrophilic residues include positively charged amino acids such as lysine (LYS) and arginine (ARG), which can form electrostatic interactions with negatively charged ligands.
Assessing electrophilicity in the active site of proteins is a fundamental aspect of drug design. It involves considering both the electrophilic nature of ligands and the nucleophilic properties of the protein's active site residues. Strategic control of electrophilic interactions has the potential to yield enhanced efficacy and selectivity in the creation of drugs for Malaria. In Figures 10 and 11, we can see the local regions involved in the stabilization into the protein pocket for the compound with higher experimental IC50.
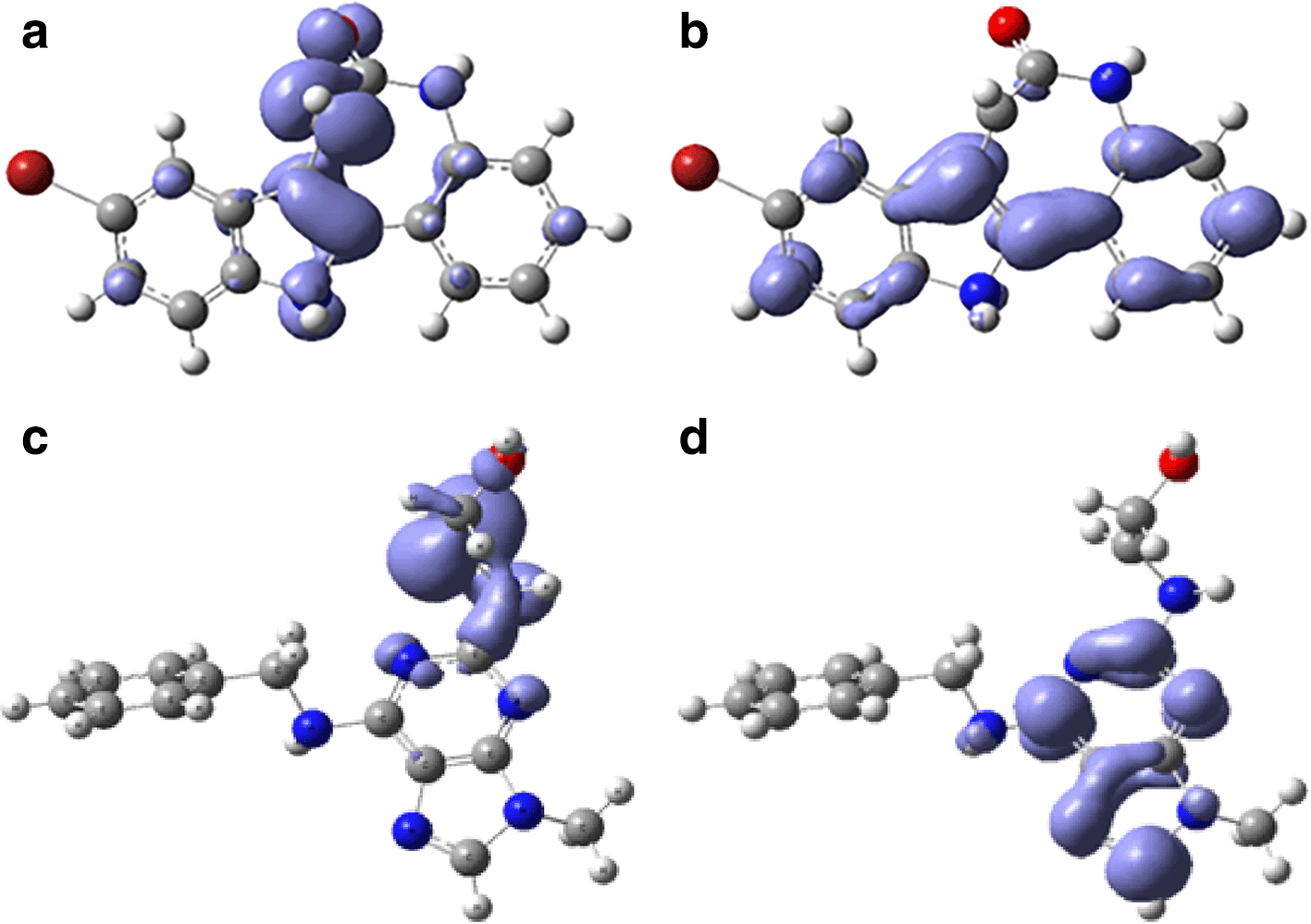
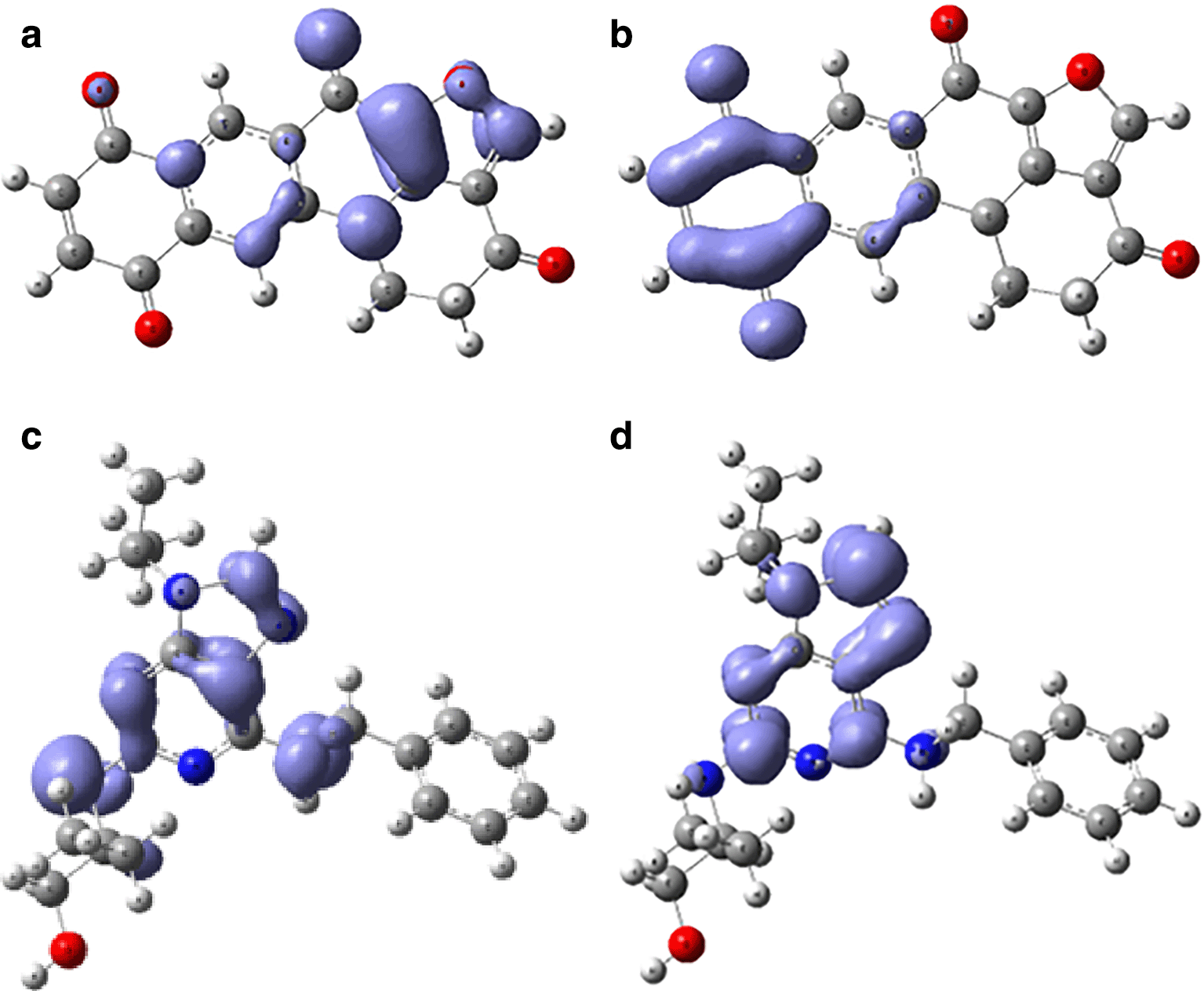
Figures 10 and 11 show good regions for the stabilization into the active site. These regions offers susceptibilities to form -H bond and consequently can be good candidates for the malaria treatment.
Molecular docking is an indispensable tool in malaria drug discovery, providing insights into the interaction between potential drug molecules and target proteins within Plasmodium parasites. The study focused on the docking interactions of various compounds, emphasizing their specific interactions with residues. Stabilizing the active site of proteins associated with malaria is crucial for designing effective and selective drugs. The docking results, shows interactions with specific residues. Stabilizing the active site enhances binding affinity, a critical factor for drug efficacy. The specificity achieved by targeting the active site minimizes unintended interactions, reducing side effects and improving drug selectivity.
Quantum Similarity Analysis, incorporating the Overlap and Coulomb operators, offers a deep electronic perspective on molecular interactions. The study presents tables indicating the electronic quantum similarity between different compounds. The analysis provides insights into chemical reactivity and potential resistance issues. The highest electronic quantum similarity values were observed between specific compound pairs, influencing the subsequent analysis of chemical reactivity. These findings prompted an exploration of global and local chemical reactivity indices to further guide drug design.
Global reactivity indices, such as chemical potential, hardness, softness, and electrophilicity, were computed for each compound. These indices play a crucial role in guiding molecular modifications to optimize drug candidates' pharmacological properties. The results identified compounds with higher chemical potential and electrophilicity, essential considerations in drug design. Understanding electrophilicity at the active site is crucial for drug design, as shown by the compound with the highest electrophilicity. The assessment involves considering the electrophilic nature of ligands and the nucleophilic properties of the active site residues. Fukui functions were employed to visualize regions involved in stabilization within the protein pocket. Figures 10 and 11 highlight specific regions susceptible to -H bond formation, making them potential candidates for malaria treatment.
In this sense, the combination of molecular docking and quantum similarity analysis, coupled with an exploration of chemical reactivity indices, provides a comprehensive approach to drug discovery for malaria.
The research identifies prospective lead compounds, underscores the significance of stabilizing the active site, and elucidates electronic considerations crucial for designing drugs that are both effective and resistant-resistant.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)