Keywords
bioinformatics, data analysis, high-throughput, transcriptomics, RNA-seq, computational workflow, reproducibility, usability
This article is included in the Bioinformatics gateway.
bioinformatics, data analysis, high-throughput, transcriptomics, RNA-seq, computational workflow, reproducibility, usability
Recent years have seen an exponential growth in new bioinformatics tools,1 a large proportion of which are dedicated to High Throughput Sequencing (HTS) data analysis. For example, there are tools to quantify the expression level of transcripts and genes from RNA-seq data,2,3 identify RNA-binding protein (RBP) binding sites from crosslinking and immunoprecipitation (CLIP) data,4 identify alternative polyadenylation sites and/or quantify their usage,5,6 or analyze single cell RNA-seq data.7 Such tools are written in different programming languages (e.g., Python, R, C, Rust) and have distinct library requirements and dependencies. In most cases, the tools expect the input to be in one of the widely used genomics file formats (e.g., FASTQ,8 BAM9), but custom formats are also frequently used. Combining such tools into an analysis protocol is a time-consuming and error-prone process. But as the analysis of HTS data (and other Big Data in the life sciences) has become such a common problem,10,11 and as the datasets continue to increase in size and complexity, there is an urgent need for expertly curated, well-tested, maintained and easy-to-use reusable computational workflows and registries to share and access them.12–14
A number of modern, feature-rich workflow specification languages and corresponding management systems,15,16 like Snakemake17 and Nextflow,18 are now gaining popularity in the life sciences, as they make the analyses easier to develop, test, share, deploy and execute. To facilitate the installation and execution of workflows across different hardware architectures and host operating systems, modern workflow management systems make use of virtualization and encapsulation techniques relying on containers (e.g., Docker19 and Singularity/Apptainer20) and/or package managers, Conda in particular. An added advantage of using workflow specification languages is that the metadata and general provenance information is stored along with the main outcomes. These may be invaluable for a more fine-grained attribution of researcher contributions, reproducibility of analysis results and insights, and cost optimization of computing resources. Ongoing work at formalizing these metadata artifacts and adding support for such standardized schemas in workflow management systems21 is very much in line with current efforts to FAIRify research data and software.22,23 All of these advantages ultimately lead to more reusable code and reproducible results, while fostering cooperativity and collaboration, both on scientific projects and on the development of Open Source Software.
A number of workflows for the analysis of bulk RNA-seq data have been developed by the community.24–30 However, out of these, some are too complex for many users to deploy, run and/or interpret, while others do not easily allow customizations and/or the analyses of large numbers of samples. Here we present ZARP, a flexible, easy-to-use workflow for bulk RNA-seq data processing that enables rapid initial insights into the data. The inclusion of the most widely used and best performing tools for the various processing steps minimizes time spent by users on making tool choices. The implementation in a widely used workflow language ensures reproducibility and reliable execution of each analysis and facilitates (meta) data management and reporting. A dedicated command-line interface tool (ZARP-cli) allows for even easier execution of the workflow and management of ZARP runs. ZARP is useful to experimental biologists who want to rapidly assess the results of their high-throughput sequencing experiments and to bioinformaticians who can not only use the preliminary results in their downstream analyses, but also adapt the analysis workflow according to their needs. Note that a preprint of this article is deposited at BioRxiv.31
ZARP (Zavolan-Lab’s Automated RNA-seq Pipeline) is a general purpose RNA-seq analysis workflow that allows users to carry out the most common steps in the analysis of Illumina RNA-seq libraries with minimum effort. The workflow is developed in Snakemake,17 a widely used workflow language,15 and follows best practice recommendations for workflow development.32 Importantly, ZARP relies on publicly available bioinformatics tools that each can be considered state-of-the-art in their specific tool class,33 and it handles bulk, stranded, single- or paired-ended RNA-seq data.
ZARP requires two distinct inputs: (i) A “sample table”, a tab-delimited file with sample-specific information, such as paths to the sequencing data (FASTQ format), reference genome sequence (FASTA format), transcriptome annotation (GTF format) and additional experiment protocol- and library-preparation specifications like adapter sequences or fragment size; (ii) A configuration file in YAML format containing workflow-related parameters, such as results and log directory paths, as well as user-related information, like a contact email address. Advanced users can take advantage of ZARP’s flexible design to provide tool-specific configuration parameters via an optional third input file, which allows for adjusting the behavior of the workflow to their specific needs. Detailed information on the input files can be found in the documentation available in the ZARP repository on GitHub.
A general schema of the ZARP workflow in its current version is presented in Figure 1. For a more comprehensive representation that includes all steps, see Figure 2. Table 1 below lists the main functionalities of ZARP and the tools that provide these functionalities, roughly in the order in which they are executed:
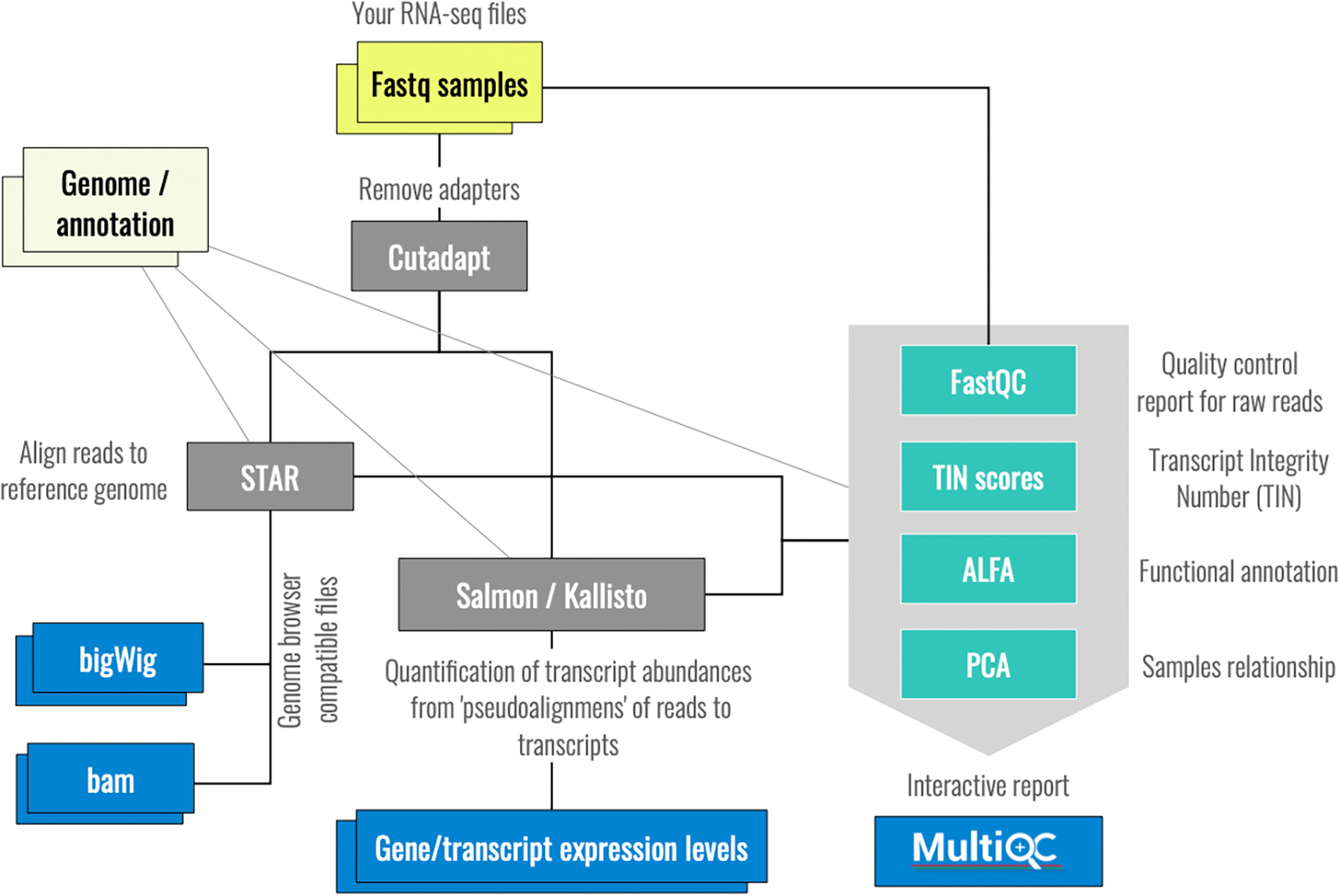
Given the input files specified in a sample table, ZARP preprocesses and maps RNA-seq reads, quantifies RNA expression levels, assesses the quality, and returns various outputs. Color legend: inputs - yellow, outputs - blue, analysis tools - gray, tools for quality control - turquoise.
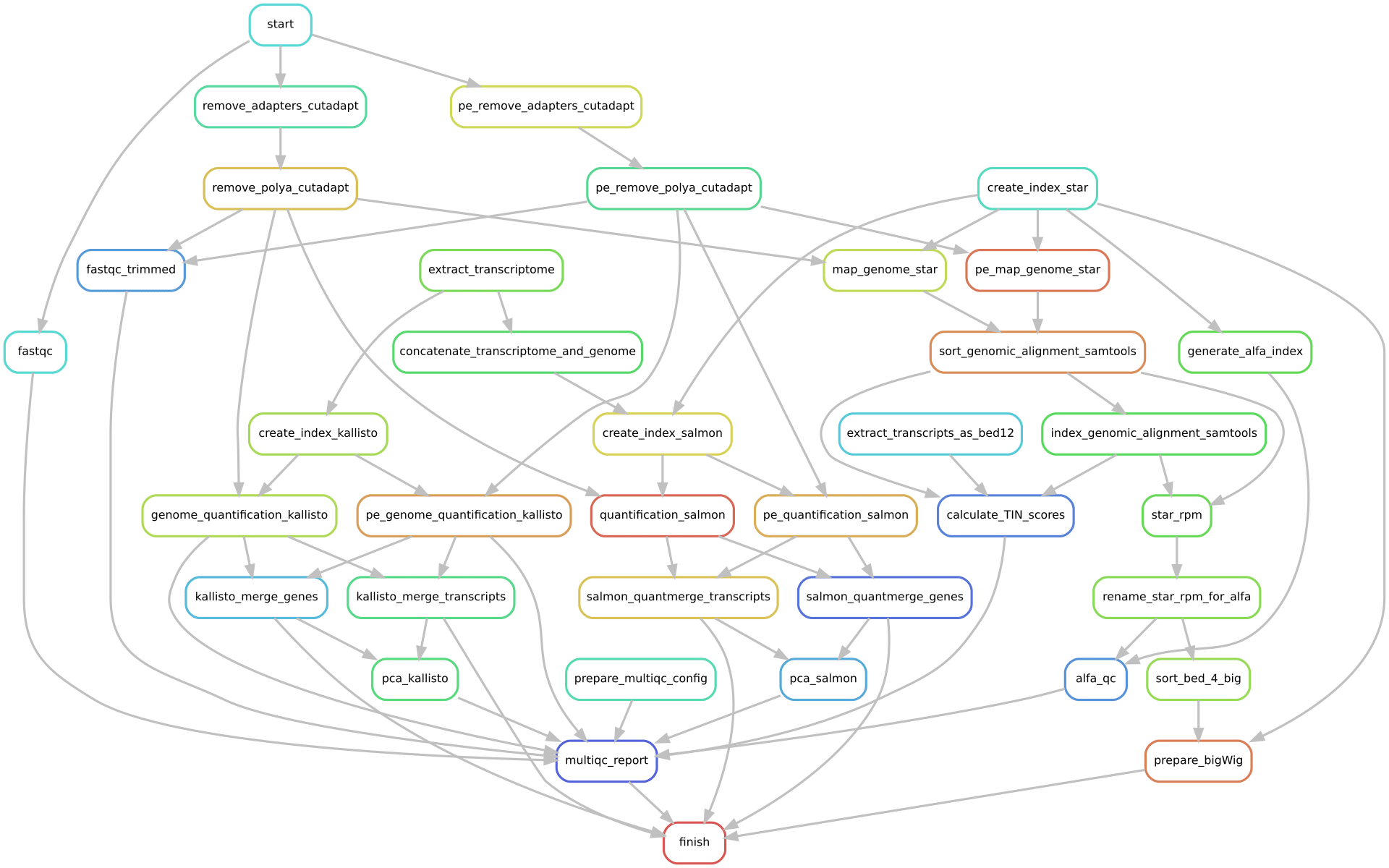
Graph-based representation of the ZARP workflow, including all of its steps (“rules”), as produced by running Snakemake with the --rulegraph option. Steps for both the single- and the paired-ended workflows are shown.
See main text for more information on use cases for each tool and why we chose those tools to be included in ZARP and ZARP-cli. Versions at time of writing are indicated.
| Tool | Description | Reference |
|---|---|---|
| FastQC 0.12.1 | Generates quality control metrics from raw FASTQ data. | GitHub |
| Cutadapt 4.6 | Trims 5’ and 3’ adapter sequences, as well as poly(A) and/or poly(T) tails/stretches. | 35 |
| STAR 2.7.11b | Aligns reads to the reference genome. | 36 |
| tin-score-calculation 0.6.3 | Calculates a transcript integrity number (TIN) for each transcript, a measure to reflect the degree of RNA degradation, based on aligned reads. | GitHub |
| ALFA 1.1.1 | Provides functional annotation information for the sample based on read alignments and gene/transcript annotations. | 41 |
| kallisto 0.48.0 | Estimates gene/transcript expression levels. | 43 |
| Salmon 1.10.2 | Estimates gene/transcript expression levels. | 44 |
| zpca 0.8.3.post1 | Performs principal component analyses of gene/transcript expression level estimates across samples included in a given workflow run. | GitHub |
| MultiQC 1.10.1 | Aggregates tool results and generates interactive reports. | 46 |
| SRA Toolkit 3.0.10 (ZARP-cli) | Fetches sequencing libraries from the Sequence Read Archive (47) and converts them to FASTQ | GitHub |
| HTSinfer 0.11.0 (ZARP-cli) | Infers sample metadata from RNA-seq libraries | GitHub |
| genomepy 0.16.1 (ZARP-cli) | Fetches genomes and gene model annotations | 49 |
Per-sample statistics obtained by applying FastQC directly to the input files (FASTQ format) provide a quick assessment of the overall sample quality. Statistics include GC content, overrepresented sequences, and adapter content. An excessive bias in GC content may affect downstream analyses and may have to be corrected for.34 Overrepresented sequences may reflect contamination or PCR duplication artifacts, which, if excessive, could lead to sparse coverage and skewed expression estimates of the transcripts of interest. Information about adapter content can help identify issues that may occur during sample preparation. For more information on the metrics that FastQC reports and how they can be interpreted, please refer to the FastQC documentation. Trimming of 5’ and/or 3’ adapters as well as poly(A/T) stretches with Cutadapt35 helps determine the proportion of informative sequences in the library and ensures a more reliable alignment of reads to a set of reference sequences to determine their biological origin. The adapters and poly(A/T) stretches to be removed need to be provided by the user as part of the sample table.
STAR36 infers the genomic origin of the reads by aligning them against an appropriate reference genome and a database of splice junctions that STAR prepares based on the corresponding gene model annotations. The reference genome and gene annotations need to be provided by the user as part of the sample table. STAR has been chosen for its very competitive performance in terms of accuracy, compared to other aligners,37 as well as for its extensive feature set and documentation. For enabling fast random access of the resulting Binary Alignment Map (BAM) alignment files, e.g., to explore them in genome browsers, ZARP then uses SAMtools9 to sort alignments by their genomic position and index them.
The sorted, indexed BAM files are further converted into the BigWig format with BedGraphtoBigWig from the UCSC tools suite.38 BigWig files allow for easy library normalization and are considerably smaller than the corresponding BAM files, thus making them convenient for visualizing and comparing coverages across multiple samples. The aligned reads are also used to calculate per-transcript transcript integrity numbers (TIN),39 a metric to assess the degree of RNA degradation in the sample, on a per-transcript basis. This is done with tin-score-calculation, which is based on a script originally included in the RSeQC package,40 but modified by us to enable multiprocessing for increased performance. To provide a high-level topographical and functional annotation of the gene segments (e.g., coding regions, untranslated regions, intergenic) and biotypes (e.g., protein coding genes, rRNA) that are represented by the reads in a given sample, ZARP includes ALFA.41
Estimating transcript abundances from RNA-seq data not only allows for subsequent differential analysis on the transcript level but also improves gene level inferences42 and is preferable to counting read alignments.2 Out of the various tools developed for that purpose,2,3 the alignment-free or “pseudoalignment”-based tools kallisto43 and Salmon44 have emerged as the state of the art for the inference of transcript abundance, due to their similarly compelling performance, ease of use, fast runtimes and memory efficiency.3 As both tools are widely used and are fairly low on resource requirements, we have decided to include both for the quantification of transcript and gene level expression. The main output metrics provided by either tool are estimates of normalized transcript/gene expression, in Transcripts Per Million (TPM),45 as well as raw read counts. For convenience, ZARP aggregates transcript and gene expression estimates across all samples with the aid of Salmon and merge_kallisto, generating summary tables that can be plugged into a variety of available tools, e.g., for differential gene/transcript expression, differential transcript usage or gene set enrichment analyses.
Within ZARP, TPM estimates are used for performing principal component analyses (PCA) with the help of zpca, a tool we created for use in ZARP, but packaged separately so that it can be easily used on its own or as part of other workflows. PCAs on gene/transcript expression levels can help users assess the relationship between samples, to detect batch effects and inform the way in which samples should be compared in downstream analyses.
Quality assessments, read alignments, genome coverages, and transcript and gene abundances can be used in downstream applications in a myriad ways, particularly when different cohorts are compared, e.g., in differential gene expression analyses. But given that such analyses require more complex inputs (e.g., experiment design tables) and are difficult to configure generically for a wide range of experiments, we deliberately decided not to include these in ZARP.
ZARP produces two user-friendly, browser-based, interactive reports: one with a summary of sample-related information generated by MultiQC,46 the other with estimates of utilized computational resources generated by Snakemake itself. Note that for the tin-score-calculation, ALFA and zpca tools, we have created plugins that enable the interactive exploration of their respective results through MultiQC.
ZARP comes with ZARP-cli, a dedicated command line interface to further simplify its usage by giving it the look and feel of a single executable.47 See Figure 3 for a general schematic outline of its functionalities and Table 1 for a list of the core tools providing them.
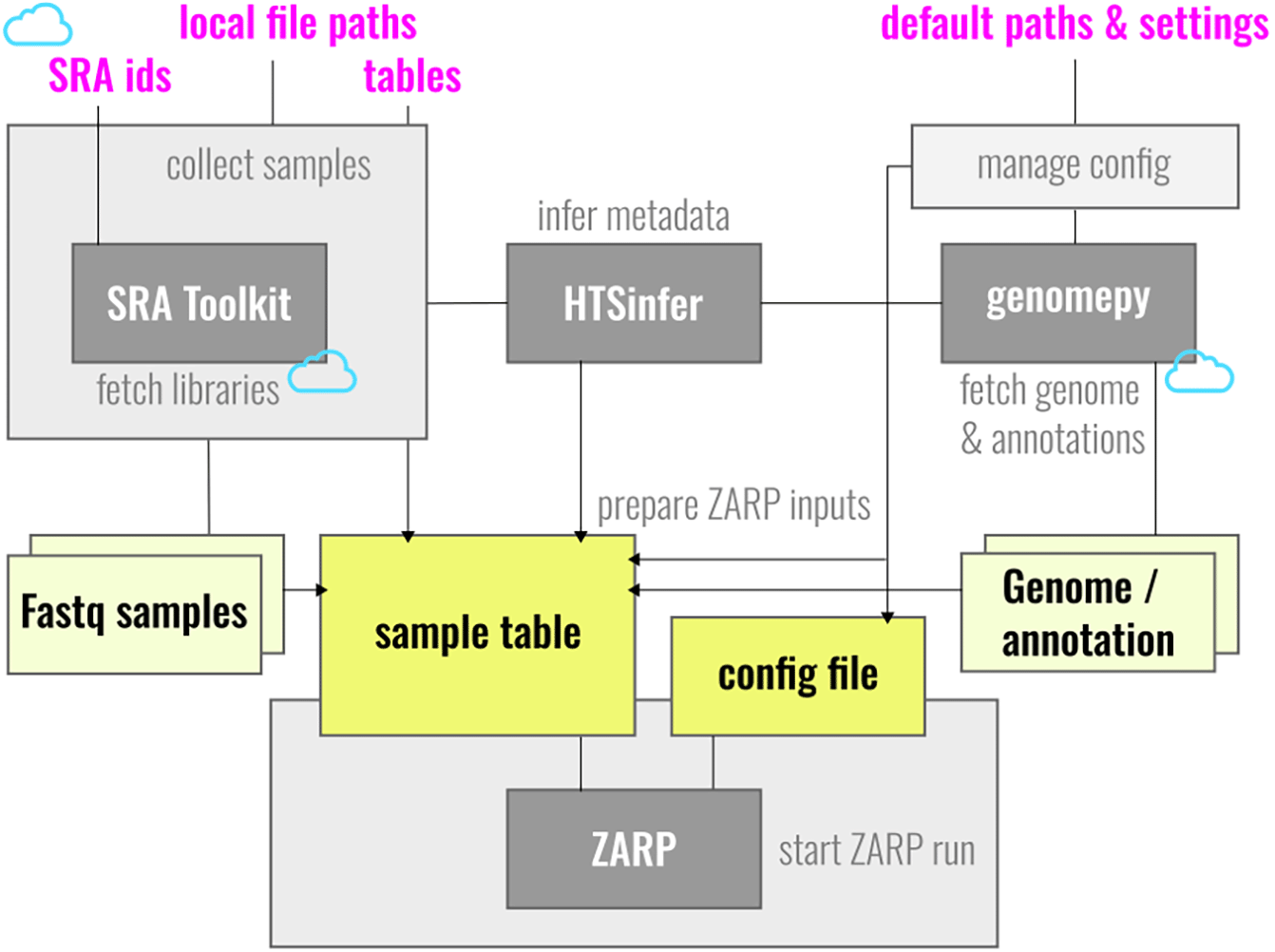
ZARP-cli consolidates different input references and compiles a sample table and a configuration file, i.e., the inputs to ZARP. Along the way, where necessary, it fetches remote libraries, attempts to infer sample metadata, including the sample source organism, and downloads the corresponding genome resources.
At the core of ZARP-cli lies its cascading configuration management: Upon first use, users set up default parameters that include sample metadata to be used if no better source is provided (or can be inferred). Configuration parameters can be overridden, on a per-run basis, with command line arguments, which in turn can be overridden, on a per-sample basis, by entries in a sample table. This design ensures that the most accurate information can be easily passed and consumed.
Once configured, ZARP-cli compiles the set of samples to analyze from the positional command line arguments specified by the user. These “sample references” can point to sequencing libraries available either locally or on the Sequence Read Archive,48 or they can point to partially or fully complete ZARP sample tables. Figure 4 illustrates all of the different ways that sample references can be passed to ZARP-cli.

Once the sample set has been assembled, ZARP-cli iteratively completes a sample table by filling in any missing information. To that effect, ZARP-cli first makes use of the SRA Toolkit to fetch any remote sequencing libraries from the Sequence Read Archive48 and convert them to FASTQ, yielding the local paths for both single- and paired-ended libraries.
Next, ZARP-cli triggers HTSinfer, a tool developed by us for the inference of RNA-seq metadata that is important for downstream analyses from the sequencing data itself. While still in an experimental/pre-release stage, HTSinfer is often able to extract, infer or assert the following: (i) whether two FASTQ files contain “mate pairs” of the same paired-ended sequencing library, (ii) the source organism from which the library is derived (out of currently more than 400 supported organisms), (iii) the relative orientation of reads with respect to the RNA transcript from which they originate and, for paired-ended libraries, one another, (iv) the frequency of commonly encountered 3’ adapters, and (v) read length statistics. Importantly, ZARP-cli uses the derived information only where such metadata is not provided by the user as part of the configuration or sample table.
Of note, both the SRA download and metadata inference steps are packaged as independent Snakemake workflows that are currently available along with the main ZARP workflow in the same GitHub repository.
After inferring sample metadata, ZARP-cli uses genomepy49 to fetch the genomes and gene model annotations from Ensembl.50 The genome version to be fetched for a given organism is pre-configured in a data file shipped with ZARP-cli; it can be easily updated or amended manually, via a text editor. To ensure consistent use of the same gene annotation version in all analyses of RNA-seq libraries within a given group, time frame or project, a default gene model annotation version can be pre-configured during ZARP-cli initialization. If no default version is set, genome resources (annotations and genomes!) are always fetched (and never reused across runs). Gene model annotation versions can also be set via a dedicated command line argument, overriding any default version set in the user configuration.
Finally, ZARP-cli assembles a sample table and a configuration file and triggers the ZARP run. In extreme cases, a ZARP-cli command can be as short as two words, zarp (the name of the program) and a single sample reference. However, making it easier to start ZARP runs is not the only benefit of using the command line interface: ZARP-cli also enforces a file and directory structure that makes it easy to find information from older ZARP runs and prevents accidental workflow re-runs for samples that have already been analyzed. Likewise, genome resources and remote libraries can be configured to be fetched only once. Finally, group- or project-specific ZARP configurations can be easily enforced, e.g., to make sure that sequencing libraries are consistently analyzed with the same set of genome resources.
ZARP development follows best practices for scientific analysis workflows.32 First of all, we chose to develop the workflow in Snakemake,17 a widely adopted workflow management system16 that allows for robust and scalable execution of computational analyses. To further enhance reusability and portability of the workflow, i.e., that it can be deployed in various scenarios, such as “on premise” High Performance/Throughput Computing (HPC/HTC) clusters or in the cloud,51 for each step (referred to as “rule” in Snakemake) of the workflow we have defined both a “rule-specific” Conda environment recipe file and a container image. The corresponding tool binaries and images are primarily hosted by the Bioconda52 and BioContainers53 registries for improved access metrics and long term preservation plans. For container images, Snakemake converts the indicated Docker images to the Singularity Image Format20 on the fly, where needed, enabling seamless execution of the workflow in environments with limited privileges (e.g., HPC/HTC clusters). Users can choose between Conda- and container-based54 execution by selecting or preparing an appropriate profile when/before running a workflow. In the current version, we include profiles only for the Slurm job scheduler. However, we will gladly accept new profiles provided by the community, and therefore we encourage users to propose the addition of their own profiles back to the original ZARP repository via raising pull requests. To reduce the storage footprint of data generated by ZARP, intermediate files can be optionally cleaned up. On the other hand, as they use a Snakemake workflow, ZARP runs generate extensive log files and workflow run metadata. This information is relevant for accounting, telemetry and provenance purposes, with current efforts ongoing at standardizing such information for improved reproducibility of workflow runs.21
The software described here follows the 4OSS Recommendations55: ZARP and ZARP-cli are hosted in their own GitHub repositories, are published under the permissive Apache, Version 2.0 free and open source software license, and follow Semantic Versioning, with each release accompanied by extensive, up-to-date documentation. We have deposited ZARP at the WorkflowHub, a registry specialized in hosting computational analysis workflows.13 It is also available in the “Standardized Usage” section of the Snakemake Workflow Catalog. ZARP-cli and any custom tools/scripts that may be useful beyond the scope of ZARP, have been largely developed according to best practice recommendations56 and deposited at Bioconda,52 which ensures that they are also available on BioContainers.53 We embrace community contributions to all of our software and try our best to ensure that all contributions are duly acknowledged in our communication. Finally, to facilitate attribution and guard against software regression and degradation, we are committed to software development best practices, such as the use of version control systems and automated continuous integration, testing and deployment workflows. In particular, for both ZARP-cli and ZARP, we provide and routinely execute extensive sets of unit and integration tests.
ZARP and ZARP-cli require a Linux operating system (tested with CentOS 8, Debian 11, 12; Ubuntu 20.4, 22.4). To install dependencies, we strongly recommend installing a flavor of Conda, e.g., Miniforge. See the installation instructions for ZARP and ZARP-cli for further details. Of note, the current ZARP version is not compatible with Snakemake 8+, because the latest Snakemake major version introduced a number of breaking changes that we are not yet supporting.
For running individual workflow steps with Conda, we further recommend installing Mamba, in line with Snakemake recommendations. Conversely, for running workflow steps inside containers, SingularityCE or Apptainer is required.
For analyzing large numbers of samples of any size and from any source, we strongly recommend running ZARP on an HPC/HTC, as the horizontal scaling and extensive multithreading will significantly reduce the wall clock time. A standard laptop or desktop machine (8 GB RAM, 4-core CPU) will be sufficient for small to medium runs (<100 samples) if only used as a Snakemake “head node”, i.e., the machine that coordinates the execution of workflow runs but leaves demanding computations to an HPC/HTC or the cloud (untested). If run entirely on a single machine, we recommend at least 32 GB (processing samples derived from certain organisms may not be possible), but preferably 128+ GB of RAM. But even then, the throughput will be limited due to reduced potential for horizontal scaling and multithreading. See section “Use Cases” below for further details on performance.
ZARP is very well suited to analyze large RNA-seq experiments or even run meta-analyses across multiple different experiments. To demonstrate how ZARP can be used to gain meaningful insights into typical RNA-seq experiments, we tested it on an RNA-seq dataset that was generated by Ham et al. (GEO accession number GSE139213) while analyzing the role of mTORC1 signaling in the age-related loss of muscle mass and function in mice.57 The dataset consists of 20 single-ended RNA-seq libraries (read length: 101 nt, gzipped FASTQ file sizes ranging from 0.8 to 3.2 Gb; library sizes ranging from 18.5 to 75.3 million reads), corresponding to four cohorts of 5 biological replicates each of muscle tissue from 3-months old mice: (i) wild-type, (ii) rapamycin-treated, (iii) tuberous sclerosis complex 1 (TSC1) knockout and (iv) rapamycin-treated TSC1 knockout. The reads were mapped against Ensembl’s50 GRCm39 genome primary assembly and corresponding gene annotations (release: 111) for standard mouse chromosomes. Other parameters for populating the ZARP sample table were mined from the metadata provided in the GEO accession entry for the sample set. Sample tables, instructions on how to start the ZARP run either via a Snakemake call or via the ZARP-cli command line interface, and select results for the test run are publicly available.58
In Figure 5, we present a subset of the outputs that ZARP generated for this dataset. One observation is the presence of slightly AU-rich reads (Figure 5A), although all samples pass the FastQC-defined threshold for GC bias, and the GC content does not exhibit a strong bias across samples. Transcript integrity across samples is also uniform and high (Figure 5B), with the highest density of expressed transcripts at TIN scores of 75 to 85. There is also no evidence of extensive sequencing of residual adapters (“adapter contamination”; Figure 5C), as less than 1% of reads in each sample have been discarded for being too short after adapter trimming. Similarly, alignment statistics as reported by STAR are also consistently high (Figure 5D), with rates of reads mapped uniquely against the mouse genome of more than 72% across all samples (<4% unmapped), irrespective of sequencing depth. As expected, ALFA analysis of transcript categories shows that uniquely mapped reads overwhelmingly originated from protein-coding genes (over 86% for all samples) (Figure 5E). Taken together, these metrics indicate that all samples are of sufficiently high quality for downstream analysis.
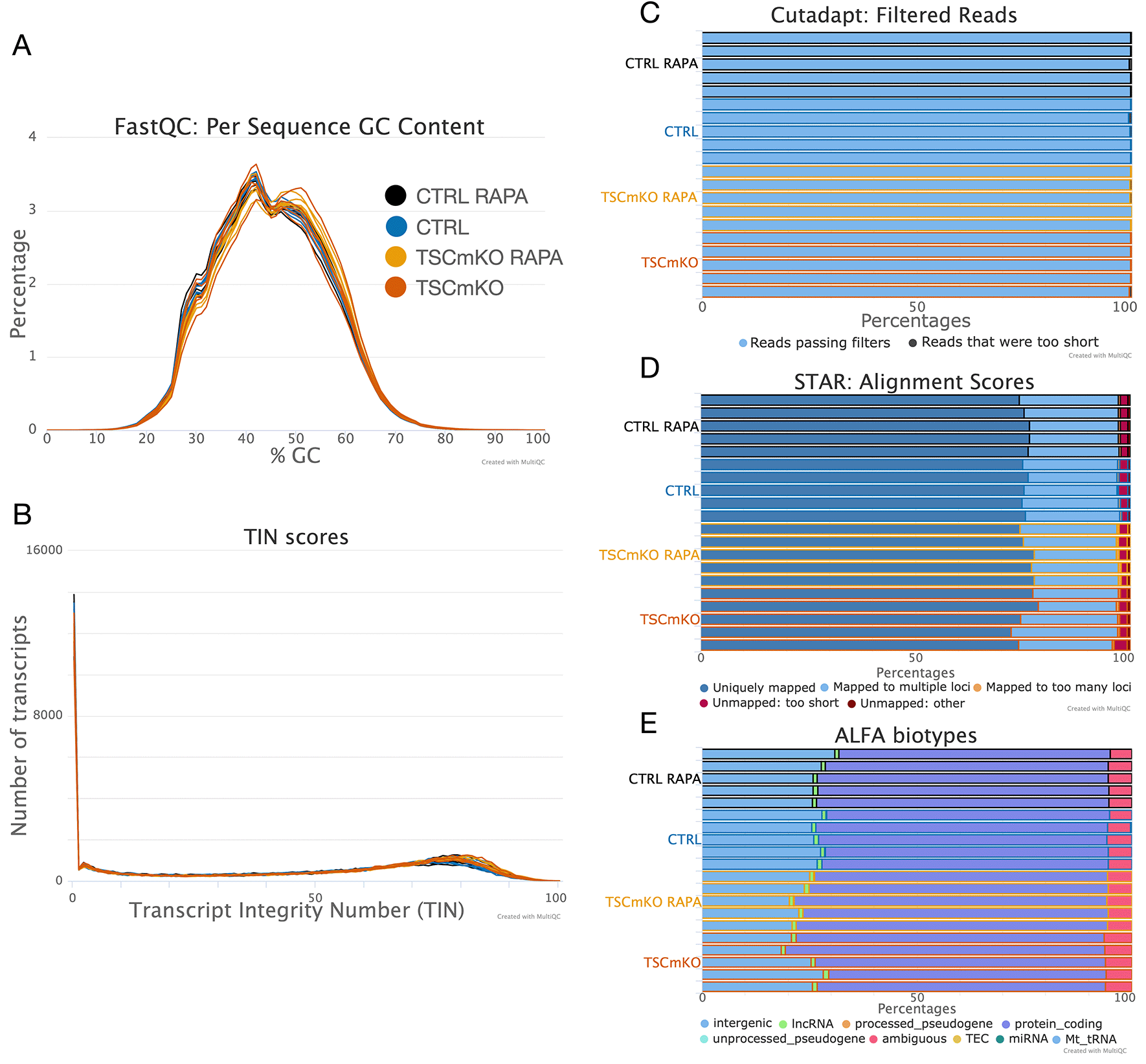
Shown are (A) GC content, (B) Transcript integrity numbers (TIN), (C) adapter removal report, (D) alignment statistics, and (E) biotypes for the test run described in the main text. Figures have been slightly edited for visibility and accessibility: some labels have been increased, cohort names have been simplified, samples have been grouped according to cohorts, and cohorts are highlighted in different colors via the MultiQC “Highlight” functionality. Additionally, some biotypes have been omitted from (E) as they are not meaningfully represented. Note that in (B), transcripts that are not expressed are assigned a TIN score of 0. The complete raw HTML report can be found at Zenodo.58 Cohort names correspond to the mouse sample cohorts described above as follows, CTRL: wild-type, CTRL RAPA: rapamycin-treated, TSCmKO: tuberous sclerosis complex 1 (TSC1) knockout, TSCmKO RAPA: rapamycin-treated TSC1 knockout.
In addition to sample-specific metrics, ZARP also performs principal component analyses across samples (Figure 6). For the test run, the distribution of samples in the space of the first two principal components shows a clustering by condition, with a clear separation between knockout and wild type, as well as between the untreated and rapamycin-treated TSC1 knockout mice. This separation is more pronounced at the gene expression level (Figure 6A), but is also present at the transcript level (Figure 6B). This shows that the differences across conditions are more pronounced than any replicate biases (multiplicative noise, sequencing errors), which strongly increases the likelihood that any subsequent analyses (e.g., differential gene/transcript expression analysis) will provide targets of biological importance.
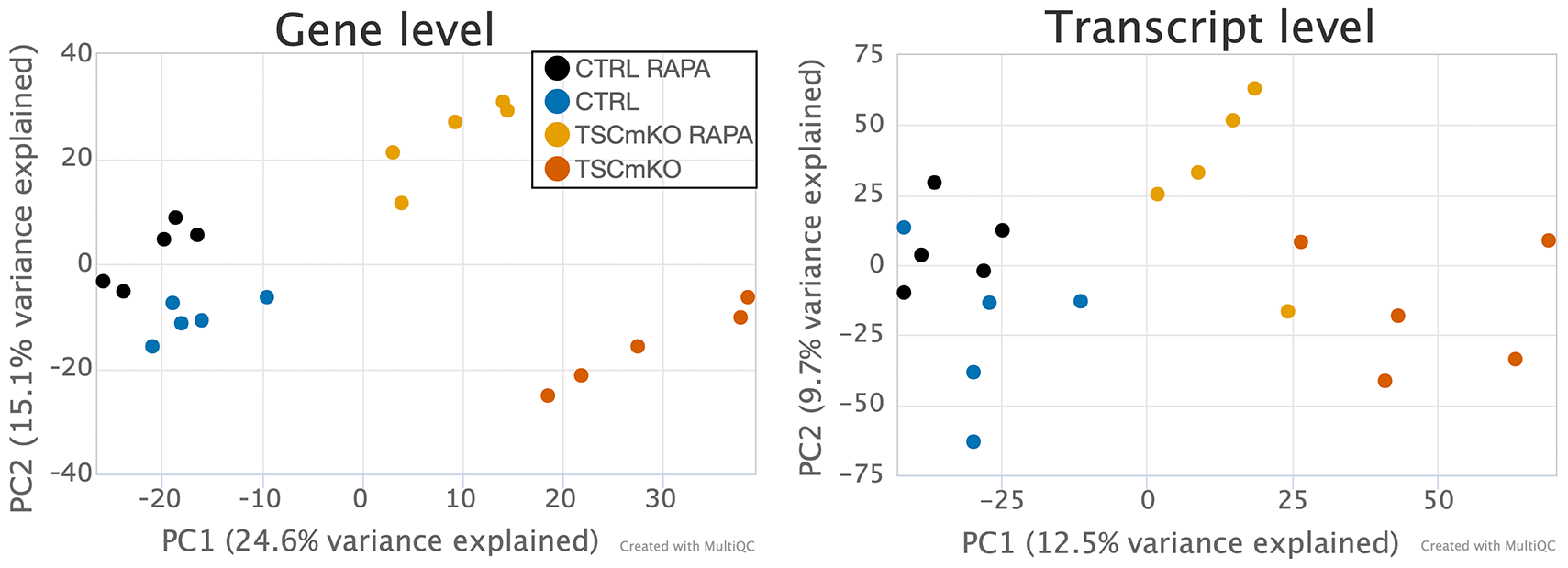
PCA at the (A) gene and (B) transcript level. PC1 and PC2 correspond to the first and second principal components, respectively. Variances explained by each of them are stated in the parentheses of the corresponding axes labels. Expression levels used in this figure are those reported by kallisto, but ZARP also generates corresponding PCA plots for Salmon-based quantifications. The figure has been slightly edited for visibility and accessibility: some labels have been increased, cohort names have been simplified and different colors for cohorts have been chosen via MultiQC’s “Highlight” functionality.
The total wall clock time to execute the entire test run was over one hour (1 h 12 min; see Figure 7) for all 20 samples on our institution’s Slurm-managed HPC cluster, where we could make heavy use of ZARP’s parallelization capabilities. This translates to a total CPU time of 71.65 h, out of which 8.32h were not sample-specific, i.e., jobs that had to be executed only once for all samples. The accumulated sample-specific CPU time used for each sample varied between 1.6h and 5h (mean: 3.2h). While the actual runtime may differ considerably across different compute environments, we project that most users would be able to run even large-scale analyses with dozens to hundreds of samples in less than a day on an HPC/HTC cluster or in the cloud, with very little hands-on time. Maximum memory usage for any of the steps and across all samples was 30.2 Gb for rule map_genome_star, which is in accordance with STAR requirements.36 With <32 Gb, this indicates that ZARP is suitable for execution on state of the art desktop and and even laptop computers, albeit at considerably higher runtimes due to the need for serial execution of individual steps and limited use of multithreading. Also, steps requiring a lot of memory are typically those that index or otherwise process genomes and/or gene annotations, so that memory requirements are higher for samples sourced from organisms with larger genomes or higher resolution annotations. None of the jobs took longer than ~35 min (wall clock time) for any of the samples (Figure 7). Among the most time-consuming steps are the creation of indices for STAR, Salmon, and kallisto (from 10min to 35 min), which, however, typically have to be performed only once per set of genome resources - and would be reused across additional runs if the same working directory is used (use ZARP-cli to enforce the consistent use of the same directory for your ZARP runs). Among the sample-specific steps, the calculation of the transcript integrity number (TIN) was the most time-consuming. However, we had already considerably reduced its runtime by adding parallelization capabilities to the original script (see Methods for further details).
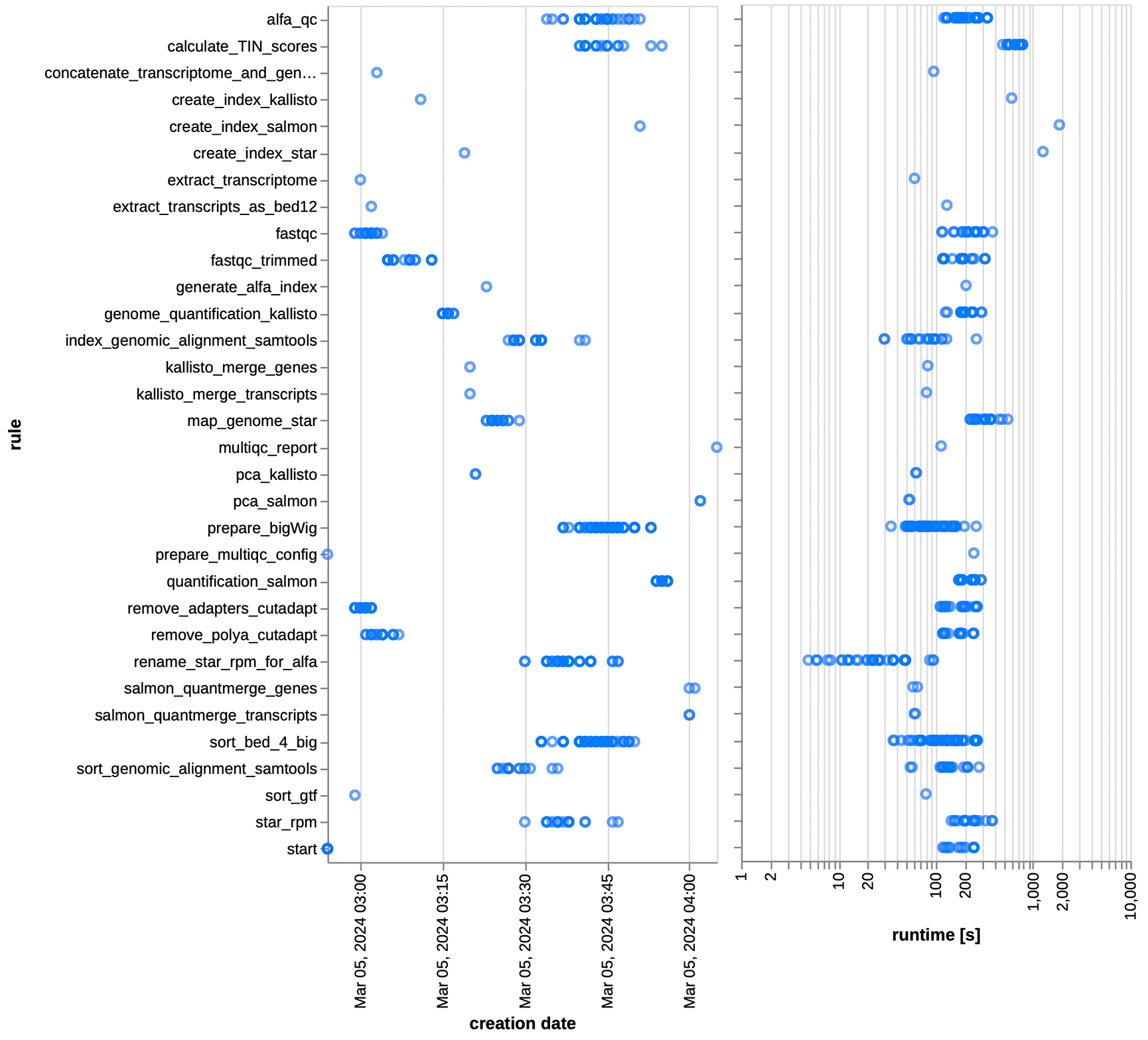
Left: Creation date of the files of each of the rules as generated by Snakemake’s reporting functionality. Right: Runtime (in seconds; wall clock time) of the different steps (“rules”) of the workflow run are depicted for each sample, as generated by Snakemake’s reporting functionality. Axis sizes were modified using Vega Editor, a functionality available directly from the Snakemake report. Differences observed across samples are a function of sample sizes, but also include sources of variation such as the time that individual jobs spent queuing on our Slurm-managed HPC cluster, as well as the hardware specifications of the cluster nodes these jobs eventually ended up on.
Triggering the test case run with an appropriate Snakemake call is easy enough, yet still requires users to fetch sample libraries and genome resources manually, from the Sequence Read Archive48 and Ensembl,50 respectively. These steps can be automated by passing the sample table to ZARP-cli instead – slightly modified to replace sample names with SRA run identifiers, and leaving the sample library and genome resource path columns empty. The modified sample table for use with ZARP-cli is also provided in our example data repository.58 Preparing a sample table is straightforward when the necessary metadata is readily available. However, in our own experience, due to a lack of widely adopted reporting standards that would reliably enable at least the most common types of downstream analyses for RNA-seq data, this is rarely the case when data is fetched from external sources. Especially for large sample sets from different cohorts, preparing a correct sample table will at the very least require painstaking literature mining. To address this issue, and to make ZARP even more accessible, we have included in ZARP-cli our RNA-seq “metadata sniffer” HTSinfer, which attempts to infer important (and widely underreported) metadata, such as the read orientation and 3’ adapters, from the sample libraries themselves. Our supplementary data repository58 includes an example of how this functionality allows ZARP users to work with a sparse sample table with parts or even all of the metadata missing, across samples from multiple organisms and prepared according to different protocols, in a single run. For quick checks, and in its most simple invocation, ZARP-cli can be called with a “two word” command like zarp SRR23590181. Examples of how ZARP-cli can be called are summarized in Figure 4.
We note that HTSinfer is still experimental, as its attempts at inferring metadata will often fail, most commonly because the data is compatible with more than one outcome and there is insufficient resolution in our inference metrics to distinguish them (e.g., a sample is compatible with two or more source organisms). Nevertheless, even the partial information that HTSinfer does provide saves substantial time on the part of the user. Moreover, ZARP(-cli) will continuously be updated with the latest version of HTSinfer to ensure that the most accurate metadata inference is made available to its users.
In summary, our test cases demonstrate how ZARP can be used to quickly gain informative insights (Figures 5 & 6) from non-trivial real-world RNA-seq datasets in a reasonable timeframe (Figure 7) and how the dedicated ZARP command line interface can be used to trigger RNA-seq analyses with unprecedented ease (Figure 4).
ZARP is a general purpose, easy-to-use, reliable and efficient RNA-seq processing workflow that can be used by molecular biologists with minimal programming experience and bioinformatics experts alike. Scientists with access to a UNIX-based computer with at least 32 GB RAM (Linux and 128+ GB RAM preferred), or to a computing cluster or commercial cloud service, can run the workflow to get an initial view of their data on a relatively short time scale. For laboratories frequently generating RNA-seq data, running ZARP on incoming data can be easily incorporated into an automated data acquisition workflow through ZARP-cli’s Python API. Similarly, ZARP-cli is particularly well suited for analyzing large-scale systematic studies that rely on hundreds or even thousands of sequencing libraries. ZARP has been specifically fine-tuned to process bulk RNA-seq datasets, allowing users to run it out of the box with default parameters. At the same time, ZARP allows advanced users to customize workflow behavior, thereby making it a helpful and flexible tool for edge cases, where a more generic analysis with default settings is unsuitable. The outputs that ZARP provides can serve as entry points for other project-specific analyses, such as differential gene and transcript expression analyses. ZARP and ZARP-cli are publicly available under a permissive open source license (Apache License, Version 2.0), and contributions from the bioinformatics community are welcome. Please address all development-related inquiries as issues at the official GitHub repositories for ZARP and ZARP-cli.
ZARP’s ease of use, coupled with its versatility, the use of state-of-the-art tools and modern technologies, its high level of adherence to best software development, Open Source and Open Science practices, and its strict focus on a well defined problem make it stand out among its competition, in particular with respect to analyzing large numbers of samples in a reproducible manner.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)