Keywords
Quorum sensing, Virtual screening, Pseudomonas, Antibiotic resistance, Docking
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Pathogens gateway.
This article is included in the Antimicrobial Resistance collection.
Quorum sensing, Virtual screening, Pseudomonas, Antibiotic resistance, Docking
Pseudomonas aeruginosa, a species of considerable medical interest, is wreaking havoc in humans due to its wide role in infections. This encapsulated, gram-negative, rod-shaped bacterium has emerged as a multidrug-resistant superbug rendering antibiotics ineffective during treatment (Lyczak et al., 2002). The opportunistic bacterium causes a plethora of diseases including cystic fibrosis, urinary tract infection, acute leukemia, endocarditis, meningitis, pneumonia, and septicemia (Bodey et al., 1983). The infections are more common in immunocompromised patients – making P. aeruginosa the second most common organism isolated in nosocomial pneumonia (17% of cases) and the third most common organism isolated in both urinary tract infection (UTI) and surgical site infection (11% of cases) (Richards et al., 1999). As a mechanism of antibiotic resistance, the bacteria can form slime-enclosed biofilms as a protective barrier (Lyczak et al., 2002). The development of biofilm is closely connected to another process termed as Quorum Sensing (QS) (Solano et al., 2014). QS is a cell-to-cell communication process through which bacteria can sense population density and regulate gene expression accordingly. Biofilm development, biofilm dispersion and upregulation of the synthesis of surfactant molecules are deeply intertwined with QS circuit (Lee & Zhang, 2014).
There are four QS systems in P. aeruginosa, namely Las, RhI, Pqs and Iqs (Scoffone et al., 2019). The Las remains at the top of the circuit in the hierarchy and is comprised of LasI and LasR – the former synthesizing an autoinducer molecule i.e. N-(3-oxododecanoyl)-L-homoserine lactone or OdDHL, and the latter receiving the OdDHL to carry out transcriptional regulation (Lee & Zhang, 2014). The binding of OdDHL to LasR initiates the cascade of signals that regulate further signaling in Rhl, Pqs and Iqs systems. The quorum sensing network in Pseudomonas aeruginosa also employs three other signaling molecules: N-butyryl-L-homoserine lactone (BHL) synthesized by the RhlI AHL (Acyl-Homoserine Lactone) synthase which binds to the RhlR receptor; 2-heptyl-3-hydroxy-4(1H)-quinolone (PSQ) binds to PqsR; and 2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde (IQS) binds to IqsR. The two receptors, LasR and RhIR, are transcriptional regulators that regulate the expression of nearly 300 genes of P. aeruginosa genome (Feltner et al., 2016). The LasR protein has two domains: ligand-binding and DNA-binding. When OdDHL binds to the LasR receptor, it binds to the ligand-binding domain and activates the protein. Consequently, the DNA-binding domain attaches to the target DNA to regulate the transcription. The production of rhamnolipids and the virulence factors such as elastase, exoprotease, pyocyanin are all regulated and influenced by the initial interaction and complex formation of LasR/OdDHL (Ochsner et al., 1995).
The search for potential quorum sensing inhibitors (QSI) has been of great interest and research showed that QSI could compete against the native ligand for binding with LasR and disrupt the QS-signaling cascade and the subsequent biofilm formation (Annapoorani et al., 2012; Zeng et al., 2008). The LasR inhibitors can be classified into three classes: non-AHL-like antagonists, AHL-like antagonists, and covalent binders (Scoffone et al., 2019). Both natural products and synthetic compounds have been researched for potential candidates. A recent study has found two potential chemical compounds that can work as quorum sensing inhibitors (Nain et al., 2019). In another study, a halogenated furanone compound from a marine alga Delisea pulchra was shown to have good anti-QS property (O’Loughlin et al., 2013). But most of the halogenated furanone is reactive and hence too toxic to be used for treatment in human (Hentzer & Givskov, 2003). There are other natural products having anti-QS activity such as patulin, penicillic acid from Penicillium species (Rasmussen et al., 2005), ajoene, a sulfur-rich molecule from garlic (Jakobsen et al., 2012), ellagic acid derivatives from Terminalia chebula Retz (Sarabhai et al., 2015), and Coumarin. Coumarin is obtained from plant extract and it has strong anti-virulence activity. It is also effective against protease and pyocyanin production and it blocks biofilm formation (Monte et al., 2014).
In our study, we used Pseudomonas aeruginosa DMC-27b (GenBank: SMRY00000000.2), a clinical strain isolated from a urine sample of an ICU patient at Dhaka Medical College hospital, Bangladesh. The original data collection took place as part of a previous study by our lab (Jahan et al., 2020), and the strain was found to be resistant against a total of 20 antibiotics with having chromosomal inheritance of all four classes of beta-lactamases (Jahan et al., 2020). The significance of using this Multi-Drug Resistant (MDR) isolate from Bangladesh offers an opportunity for a case study and investigate into any possible mutation in the LasR protein that might have an impact on the ligand binding modes. Considering the emergence of new MDR Pseudomonas. aeruginosa strains across the globe, and how south Asian countries like Bangladesh are acting as epicenters of AMR (Antimicrobial resistance) spread, it is critical to survey changes in the LasR receptor and continue to enrich the pool of new inhibitors.
The cheminformatics analyses were carried out in Schrodinger Maestro v11.8 (Schrodinger, LLC, New York, NY. There is a free academic version of this license that can be used to replicate this work. In addition, readers can use AutoDock Vina as an alternative software that carries out a similar function). This study utilised a desktop PC with Intel (R) Core™ i7 7700/4.0GHz processor, 8GB DDR4/3200 MHz RAM, and 4GB AMD Radeon RX 570 graphics with support for OpenCL 2.0 running Windows 10 Professional operating system. The entire workflow was segregated into three stages (Figure 1). Stage-I included genome retrieval and annotation, gene profiling, homology modeling of protein, and database search for AHL-like compounds. The stage-II included the protein and compounds preparation leading to molecular docking, molecular mechanics based binding free energy calculation. Stage-III began with further analysis comprising of ADMET analysis, induced fit docking and molecular dynamics. Finally, the lead compounds were comparatively analyzed with respect to the protein from the reference strain.
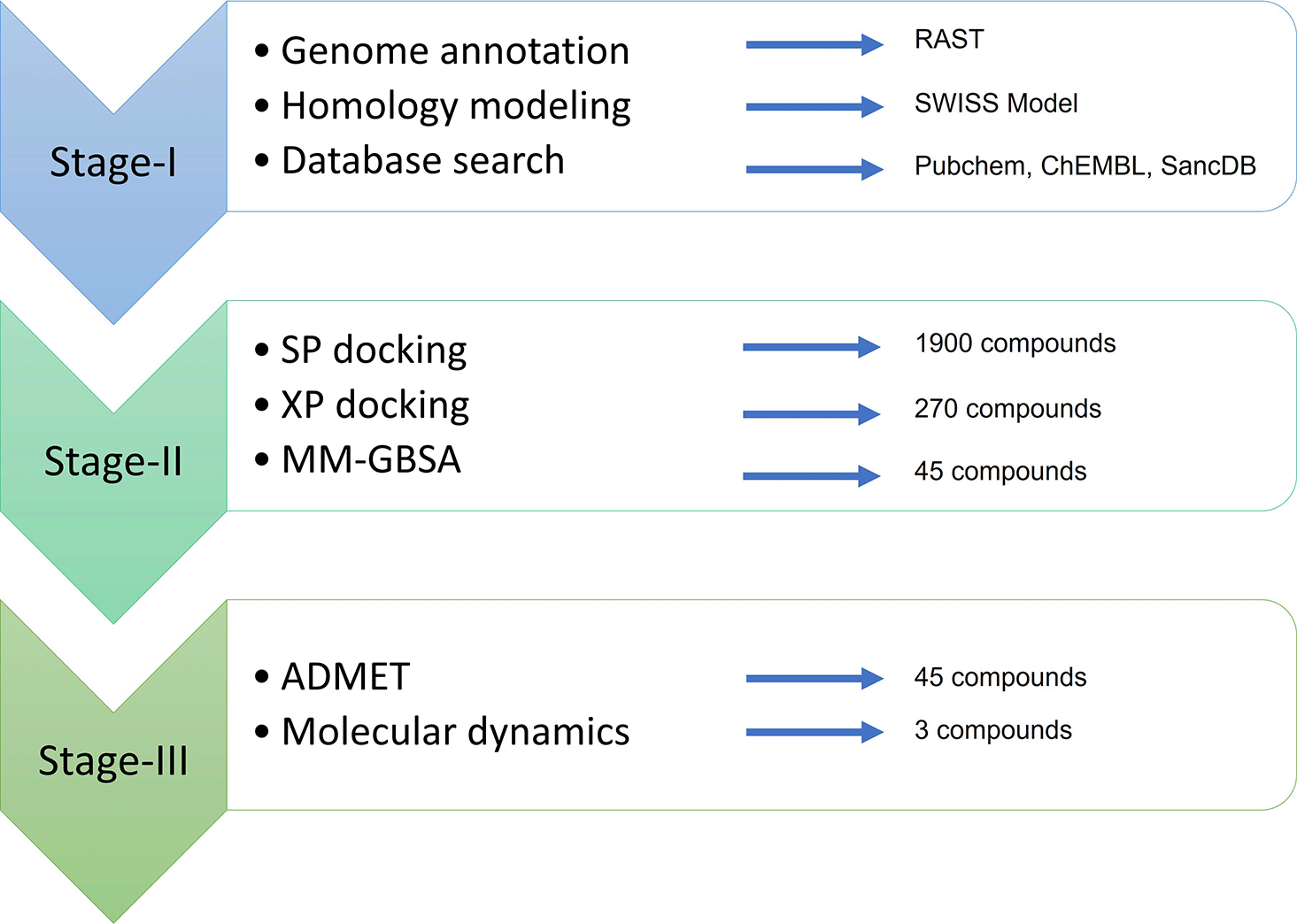
SP, Standard Precision; XP, Extra-Precision; MM-GBSA, Molecular mechanics-Generalized Born Surface Area; ADMET, absorption, distribution, metabolism, excretion, toxicity; RAST, Rapid Annotation using Subsystem Technology.
The genomic data were retrieved from National Center for Biotechnology Information (NCBI) for the strain Pseudomonas aeruginosa DMC-27b with the GenBank accession number SMRY00000000.2 (Pseudomonas Aeruginosa Strain DMC-27b, Whole Genome Shotgun Sequencing Project, 2019). The raw data was uploaded to the publicly available web server RAST (Rapid Annotation using Subsystem Technology) for automatic annotation. The presence of LasI/LasR and the proteins involved in the QS system were confirmed by RAST and were furthered validated by custom Basic Local Alignment Search Tool (BLAST) searches inside the RAST SEED viewer version 2.0 (Overbeek et al., 2014).
The amino acid sequence of the LasR protein of P. aeruginosa DMC-27b was submitted to the publicly available SWISS-MODEL web server for homology modelling (Waterhouse et al., 2018). The protein template PDB: 3IX3 was selected owing to its high resolution (i.e. 1.4 Å), standard range R-free value (i.e. 0.207) and an excellent score in Ramachandran outliers (i.e. 0.0%). The generated 3D homology model (created using SWISS-MODEL) was created in homodimer containing two chains: A and B and both contained the co-crystallized ligands. The quality of the resulting protein structure was validated using QMEAN-Z score and Ramachandran plot analysis.
The original ligand (i.e. N-3-Oxo-Dodecanoyl-L-Homoserine Lactone) for the LasR protein was used to observe the protein-ligand interaction, and to identify the core structure of the ligand. A total of three chemical databases were used to download chemical compounds. PubChem (Kim et al., 2019) and ChEMBL (Davies et al., 2015) were used to find and download chemical structures similar to N-3-Oxo-Dodecanoyl-L-Homoserine Lactone. Next, a complete SancDB database was downloaded (Hatherley et al., 2015) in the form of SANCDB SDF structure PubChem is a massive open repository of experimental data and ChEMBL is a manually curated chemical database of compounds with drug-like properties. SancDB, which stands for South African Natural Compounds Database, is a database containing compounds isolated from the plant and marine life in and around South Africa. PubChem and ChEMBL returned 1080 compounds, and SancDB returned 820 compounds totaling the number of compounds to reach 1900 - see Underlying data (Siam, 2023) for the full list (re-uploading permissible under copyright).
The protein model (LasR_DMC27b.pdb, generated using SWISS-MODEL by keeping 3IX3 as a reference) was not suitable for molecular docking in its unprepared state (Kalia et al., 2017) because an unprepared protein structure may contain gaps, missing atoms, and other structural irregularities that may affect the accuracy of the docking simulation. The protein structure was prepared by Schrodinger’s Protein Preparation Wizard (part of Schrodinger Maestro). During preparation, proper bond orders were assigned, hydrogens atoms and missing residues were added, water molecules beyond 5.00 Å distance were removed from hetero groups, and hetero states were generated using Epik at pH: 7.0 ± 2.0. The protein was further refined by optimizing the H-bond assignment and running restrained energy minimization of the structure using force field Optimized Potentials for Liquid Simulations (version OPLS_2005) until the heavy atoms converged to the RMSD value of 0.30 Å (Banks et al., 2005).
All the selected 1900 compounds were downloaded in their three-dimensional structure-data file (SDF) format. A phase database was created using Schrodinger Maestro v11.8 to assemble the potential ligand-like compounds. The Schrodinger LigPrep v2.2 module (part of Schrodinger Maestro) was applied to generate tautomers and ionization states at pH: 7 ± 2.0 and to minimize energy using force field OPLS_2005. The Epik v4.6 (part of Schrodinger Maestro) was used to compute the predicted pKa values for drug-like molecules; and reactive functional groups were eliminated from the result which might give false positive during screening (Shelley et al., 2007). To filter the unsuitable compounds, Schrodinger QikProp module (part of Schrodinger Maestro) was applied to test the Lipinski’s Rule of five (Lipinski et al., 2001) and Jorgensen’s rule of three (Jorgensen, 2004).
The grid-box was generated using the Schrodinger Glide v8.1 module (part of Schrodinger Maestro) based on the co-crystallized ligand structure in LasR-DMC27b PDB model. For flexible docking, the active site was confined to a larger enclosing box in proportion to the defined smaller box (27×27×27 Å) around the potential binding site of interest using the centroid native ligand. The Van der Waals radius scaling factor was set to 1.0 Å and the partial charge cutoff was kept at 0.25. There was no constraint or rotatable group set during the grid-box generation.
The Schrodinger Glide v8.1 was used to run Standard Precision (SP) docking for 1900 compounds employing the generated receptor grid (Friesner et al., 2004). Based on the literature review, an initial cutoff value of -7.5 kcal/mol was considered for filtering which returned 270 compounds (Nain et al., 2019). For the second phase, the compounds were docked using XP (Extra Precision) method and this time, the cutoff value was set to -8.75 since the native ligand (OdDHL) binds to the LasR_DMC27b with an XP score of -8.72 kcal/mol. In both cases, the flexible ligand sampling scheme was used. Compounds with more than 500 atoms and 1000 rotatable bonds were skipped and Epik state penalty was added to the docking score. In post docking minimization, the number of poses per compound was set to five. After XP docking, a total of 45 compounds were assessed comprising of different variations of PubChem compounds: K5D, K5G, K5J, OHN, and 443433.
The Schrodinger Prime v3.3 [Prime, Schrödinger, LLC, New York, NY, 2019], part of Schrodinger Maestro, was used to calculate MM-GBSA (Molecular Mechanics-Generalized Born Surface Area) based End-Point Binding Free Energy Calculation, which is an algorithmically rigorous, computationally intensive and superior to molecular docking. The MMGBSA methodology yields a credible assessment of binding free energy, expressed in Kcal/mol units, and may serve as a suitable approach for post-docking validation purposes. Here, GBSA continuum solvent model, as outlined in the work of (Lyne et al., 2006), was utilized to conduct simulations on a set of 45 compounds. Schrodinger Prime incorporates a surface generalized Born (SGB) model that utilizes a Gaussian surface in place of the Van der Waals surface to enable a more accurate representation of the solvent accessible surface area. Subsequent to the docking procedure, the binding free energy (ΔGbind) for each compound was calculated. Schrodinger Prime uses a surface generalized Born (SGB) model employing a Gaussian surface instead of Van der Waals surface for better representation of a solvent accessible surface area. Based on the docked complex, the binding free energy (ΔGbind) of each compound was calculated. All 45 compounds showed lower binding free energy than the native ligand, thus validating the result of XP docking.
The 45 selected compounds were tested for drug-likeness and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties using the Schrodinger QikProp v5.8 module (Ioakimidis et al., 2008). The druggability prediction is a crucial step in filtering the compounds that might be unsuitable for use in the human body. Unlike fragment-based approaches, QikProp’s predictions are based on the full 3D molecular structure, providing accurate results on par with the properties of the 95% of known drugs. The QikProp’s result yielded 44 properties for the chemical compounds, of which, ADMET properties and molecular descriptors were taken into consideration particularly octanol/water and water/gas log Ps, log S, log BB, overall CNS activity, Caco-2 and MDCK cell permeabilities, log Khsa for human serum albumin binding, HumanOralAbsorption and log IC50 for HERG K+-channel blockage. Since most halogenated furanone compounds are too reactive for use as drugs for their high electrophilicity, they were carefully removed from the result letting only 3 compounds to pass through the filters based on the QikProp’s results.
The stability and intermolecular interactions between protein and molecules were investigated using molecular dynamics (MD) simulation (Liu & Kokubo, 2017) techniques with a 100 ns time interval using the complexes obtained from docking studies to further investigate the protein and ligand binding mode. MD simulations were run with the Desmond module from the Schrödinger Release 2020-4 (Academic edition) suite (Molecular dynamics can also be done using GROMACS which is free to use). A TIP3P water model (density: 0.997 g/L) at physiological conditions (298 K, pH 7.4) with an orthorhombic box shape as the boundary was used to represent the complex protein-ligand interaction (Mark & Nilsson, 2001). On all three axes, the minimum distance between the protein surface and the boundary was set to 10 Å. The system was neutralized by adding 0.15 M Na+ and Cl- salt concentrations (Paul et al., 2023). Before simulation, NPT parameters (constant particle number (N), pressure (P), and temperature (T)) are utilized representing an isothermal-isobaric ensemble, which simulates the most typical experimental conditions. The MD simulation was run with the OPLS-2005 force field at constant pressure (1.01325 bar) and temperature (310 K), with recording intervals of 100 ps. These simulation conditions were set to mimic human cell microenvironment. The Simulation Interaction Diagram (SID) of the Desmond module of the Schrödinger program was used to analyze the trajectories acquired from the MD simulation. Based on the simulated trajectories, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and Protein-Ligand interactions were utilized to assess the stability of the ligand-protein complexes.
A comparative docking analysis between our laboratory strain and the NCBI reference strain was done. The LasR_DMC27b.pdb was obtained from Pseudomonas aeruginosa DMC27b and the reference protein 3IX3 was obtained from Pseudomonas aeruginosa PAO1 and downloaded from the Protein Data Bank (PDB). The selected inhibitors were used against the reference protein 3IX3. Molecular docking and MMGBSA were performed in case of both proteins using the selected compounds and the native ligand for comparison.
The quorum-sensing circuit in P. aeruginosa DMC-27b showed substantial resemblance to P. aeruginosa PAO1 except for RhlL and PhzD proteins. All the required proteins responsible for QS were found to exist in P. aeruginosa DMC-27b indicating capacity for quorum sensing and biofilm formation. The presence of LasI, LasR, RhlR, RhlI and RhlL were manually confirmed by BLASTp search. The activation of the Las system further activates the Rhl system that produces rhamnolipids for which the contributing proteins (RhlI, RhlR, RhlA, RhlB, RhlC, RhlE, RhlL, RhlG) were all present in the P. aeruginosa DMC-27b. A notable difference was found in Rhl system as P. aeruginosa DMC-27b had both Rhl-I and Rhl-L proteins that contribute to N-butyryl-L-homoserine lactone synthesis and N-acyl-L-homoserine lactone synthesis, respectively. This property might provide the bacteria with an evolutionary advantage over the reference strain (PAO1) as the Rhl system can make use of quorum sensing signals by generating both types of homoserine lactones. Similarly, the absence of PhzI in the phenazine system indicates that the PhzR could be co-activated by different acyl-homoserine lactones. The production of exoprotease (encoded by lasA), elastase (encoded by lasB), biofilm dispersal mediator (C-di-GMP binding protein), the regulator of hydrogen cyanide synthase (ANR) were also present that play role in virulence. In the Phz system, PhzR was present, but the PhzI was not identified.
The quality of the resulting protein homology structure (denoted here as LasR_DMC27b) was checked and the QMEAN-Z score was 0.99 and the all-atom score was 1.92. One mutation was reported in the ligand-binding domain at the position of amino acid no. 171 (Glycine instead of Valine) and it was further confirmed by way of EMBOSS matcher 6.6.0. The LasR protein has two domains: amino-terminal ligand-binding domain and a carboxy-terminal DNA-binding domain. The mutation in our strain is at the position of amino acid no. 171 (Glycine instead of Valine) at the terminal region, far away from its natural active site, hence it might not have considerable effect on the ligand-binding activity as seen from our study. The solvation and torsion scores were within an acceptable range. In Ramachandran Plot analysis, the number of residues in the favored region was 319 (99.1%); the number of residues in the allowed region was 3 (0.9%); and the percentage of outlier was 0.0% (Figure 2).

(A) In Ramachandran Plot, about 99% of residues are in the favored region with 0.0% outliers. The image is generated and viewed using SWISS PDB viewer. (B) the Pairwise Alignment between LasR_DMC27b & 3IX3 has Identity: 171/172 (99.4%), Gaps:1/172 (0.6%), and Score: 892; performed using EMBOSS matcher 6.6.0.
Three compounds were obtained as potential inhibitors denoted as L1, L2 and L3 after sequential identification process in order of- molecular docking, MMGBSA, molecular dynamics, ADMET prediction and were further improved with induced fit docking.
There was slight difference in the SP and XP docking scores of the selected compounds. All three compounds (L1, L2. L3) showed higher scores in SP, XP docking compared to the native ligand (OdDHL). The molecular docking result is shown in Table 1. The extra precision (XP) docking score for the OdDHL was found to be about -8.7 kcal/mol which is on par with other studies (Nain et al., 2019). All three selected compounds showed better binding capacity against both the clinical strain (i.e. DMC27b) and the reference strain (i.e. PAO1). The potential compounds showed greater free binding energy than the native ligand and had reasonable ligand strain energy. The compound L2 and L3 had variation in ligand strain energy, 5.198 kcal/mol and 8.041 kcal/mol, respectively, despite having a very similar nature of chemical structures.
IUPAC, The International Union of Pure and Applied Chemistry; SP, Standard Precision; XP, Extra-Precision.
In the binding mode analysis, the selected compounds were well within the protein’s active site. Compounds no. L1, L2 and L3 showed similar patterns of hydrogen bond interaction (Figure 3). The potential quorum-sensing inhibitors (QSI) were bound to the protein’s active site by way of hydrogen bonds to the amino acids Tyr56, Ser129, Asp73 and Trp88. This showed a crucial difference in comparison to the native ligand (OdDHL) which binds to the protein using Tyr56, Ser129, Asp73 and Trp60 (Table 1). Furthermore, LasR can bind an array of compounds containing the lactone head ring. Furthermore, the primary binding site for OdDHL is a combination of hydrophilic and hydrophobic interactions where the lactone head group facilitates hydrogen bonds stabilizing the structure while the acyl tail contributes to hydrophobic interactions. Reports show that LasR has two binding modes: one that mimics the canonical autoinducer binding arrangement, and the other with the lactone head group rotated approximately 150° (Paczkowski et al., 2019). The selected three compounds lack the lactone head group but facilitate strong hydrogen bonds with the receptor. All the compounds hydrogen-bonded with Tyr56, Ser129, Asp73 and Trp88. The tyrosine-56 and Serine-129 are the two most defining amino acid residues for LasR binding to its ligand. But, the native ligand binds with the receptor using Tyr56, Ser129, Asp73 and Trp60 instead of Trp88 found in the L1, L2 & L3 inhibitors. This difference in the binding mode of the potential inhibitors could play a key role in inhibiting the quorum sensing signaling.

(A) Compound L1 hydrogen-bonded with Tyr56, Ser129, Asp73, Trp88 residues of the LasR receptor. Similarly, (B) Complex-L2 showed HB interactions with Tyr56, Ser129, Asp73, Trp88 residue, and (C) Complex-L3 shared HBs with Tyr56, Ser129, Asp73, Trp88 residues, and (D) the reference compound shared HBS with Tyr56, Ser129, Asp73, and Trp60 residues.
In the MMGBSA analysis, the three compounds showed lower binding free energy than the native ligand (Table 2). The free energy for the native ligand (OdDHL) binding to the receptor LasR was -115.08 kcal/mol, and the binding free energy for L1, L2 and L3 ranged from -128.65 kcal/mol to -127.51 kcal/mol which is lower than OdDHL. The obtained coulomb energy affinity was significantly higher than the native ligand, -65.27 kcal/mol to -74.75 kcal/mol versus -33.9 kcal/mol, respectively. The compound L1 had the lowest ΔGBind energy, but compound L2 had comparatively better coulomb energy as well as the solvation energy of the complex. The native ligand had more ligand strain energy (SELig) than the selected compounds.
All three compounds retained satisfactory results with values in the range of 95% of known drugs. The ADMET properties of the selected compounds were within the acceptable range for optimal drug candidates. None of the compounds violated Lipinski’s rule of five or Jorgensen’s rule of three. The HERG value, central nervous system (CNS) activity, binding to human serum albumin and other properties were within limit. The human oral absorption of the compounds was above 80%. The QPlogHERG values for L1, L2 and L3 were -4.964, -4.59, -3.688 respectively. A value below -5.00 raises concern as to whether the compound might act as hREG inhibitor for K+ ion channel. Since the L1 compound is close to the threshold level for QPlogHERG value, it must be experimentally checked for hREG inhibition activity. The human oral absorption rate for all three compounds was excellent (>80%) and the compounds did not show acute oral toxicity. The ADMET profile of selected three compounds is provided in Table 3. Furthermore, the molecular descriptors for the compounds were analyzed and shown in Table 4.
No suitable inhibitor was found from ChEMBL and SancDB databases. Despite some compounds having good XP score, all of them were eliminated in the ADMET screening stage. The chemical structures of the L1, L2, L3 and OdDHL are presented in Figure 4. The compounds lack the ester functional group in the ring implying that there is no lactone headgroup which is the characteristic of the native ligand, OdDHL.

The L2 and L3 chemical compounds have similar chemical structures with one ring while L1 contains two rings. All selected compounds lack the characteristic lactone headgroup of OdDHL.
Molecular dynamics were run using the LasR_DMC27b protein and the three selected ligands as well as the reference ligand, OdDHL. After 100 ns simulation period, the ligand-protein RMSD (root mean square deviation) results indicated that all three complexes were close to the permissible limit of 3.0 Å with respect to the reference ligand complex. However, complex-3 had a value >4.0 Å during the 20 ns and later at 95 ns simulation period (Figure 5(A)) and it was not equilibrated until 50 ns. All the complexes were equilibrated after 30 ns and found to be stable, albeit the reference complex was not found to be equilibrated at the beginning but after 80 ns, it had the lowest RMSD, indicating lesser fluctuation and improved stability. Complex-1 has shown superior stability and is closer to the reference complex than the other experimental complexes. In terms of the LasR-DMC27b protein’s stability, all three selected ligand-protein complexes were quite stable as they showed an RMSD value lower than 3 Å. Compared to all, the complex-2 showed better protein stability (Figure 5(B)). The conformation changes occurring to the protein’s side chain were analyzed using the root mean square fluctuation (RMSF) values. Greater fluctuations are usually found in the protein’s N-terminal and C-terminal domains, and this was also the case for the LasR-DMC27b protein and the docked complexes. Except for a few spikes, the complexes exhibited low fluctuations overall indicating their potential to be used as antagonist compounds (Figure 5(C)).
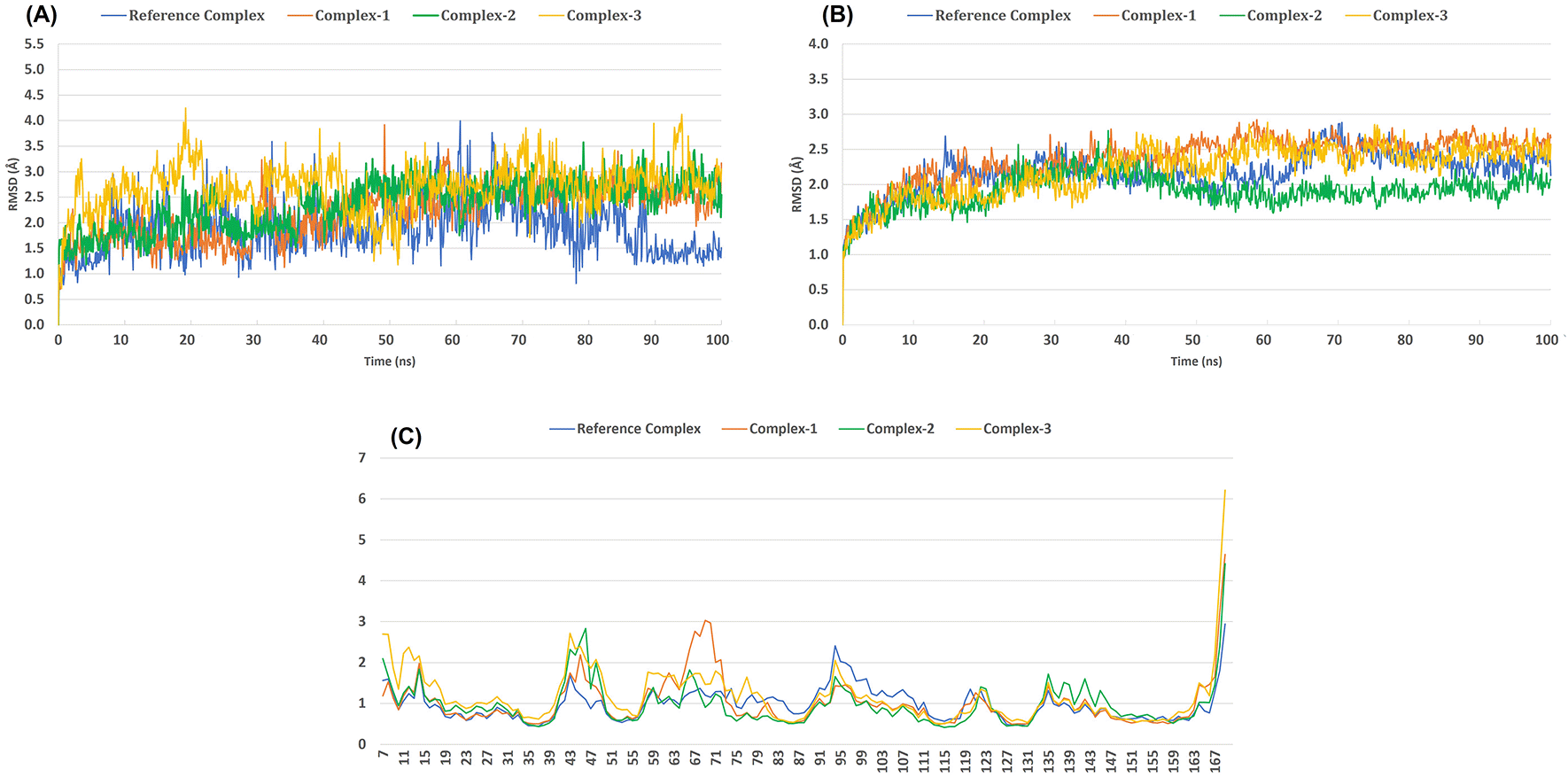
(A) Ligand-protein complex RMSD (root-mean-square deviation), (B) Protein Cα RMSD and (C) Protein Cα RMSF (root-mean-square fluctuation) of the backbone-atoms of the docked LasR-DMC27b protein.
Using the default parameters of the Desmond module, the MD trajectories of the LasR protein, three possible lead compounds, and the reference complex were analyzed. In protein-ligand interactions, Complex-1, Complex-2, and Complex-3 hydrogen bonded with residues including ASP_73, THR_75, THR_115 and SER_129, while reference complex further formed hydrogen bonds with TYR_56 & TRP_60. Moreover, complex-3 formed hydrogen bonded with TYR_64, TRP_88 & TYR_93 exclusively. There was a weak ionic bond with ASP_73 in complex 3. There were hydrophobic bonds and water bridges present in various residues in all the complexes (Figure 6).
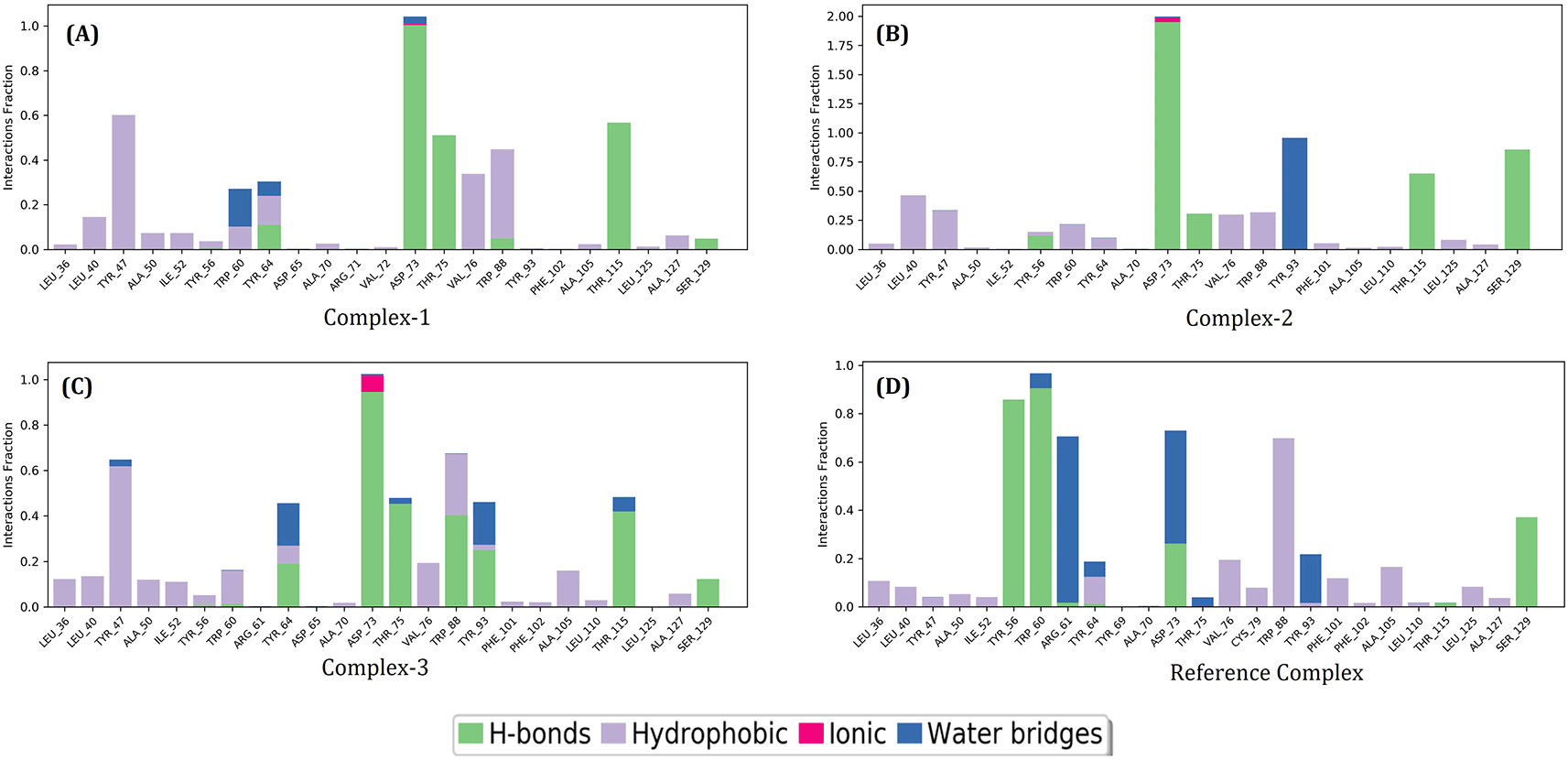
In the post-molecular dynamics MMGBSA analysis, out of the three compounds, L2 compound showed lower binding free energy (ΔGBind = -88.97 kcal/mol) than the native ligand (Table 5). The ΔGBind free energy for the native ligand (OdDHL) binding to the receptor LasR was -80.58 kcal/mol, and the binding free energy for L1, L2 and L3 compounds ranged from -69.7 kcal/mol to -88.97 kcal/mol. This is lower compared to MMGBSA analysis prior to molecular dynamics which ranged from -128.65 kcal/mol to -127.51 kcal/mol. The compound L2 also had the lowest hydrogen bond energy of the complex as well as the lowest Coulomb energy of the complex. This indicates that L2 compound was a better drug candidate compared to L1 and L3.
Our study reported no major mutation in the LasR protein in P. aeruginosa DMC27b strain that could make it functionally different from the reference strain. We further performed a comparative analysis between the two strains to identify if docking score changes between two proteins due to single amino acid mutation in the terminal region. Our comparative analysis showed that the selected three compounds (L1, L2, and L3) had consistently better binding capacity to both receptors than the native ligand OdDHL. This finding further strengthens the hypothesis that ligand screening in single isolate could be relevant for use in other strains. A further in-depth study could be performed to observe how inhibitors reported by other studies would act against LasR across different strains of P. aeruginosa. Table 6 shows the comparison of interaction of selected compounds and the native ligand with both receptors, LasR_DMC27b and 3IX3.
XP, Extra-Precision.
Our study has its limitations: first, the research design was based on similarity search to find potential ligands, and this has its inherent limitation that potential inhibitors must be further chemically synthesized and experimented in-vitro for efficacy. Second, we could not perform longer than 100 ns simulation in molecular dynamics due to resource constraints. However, the selected compounds in this study were found to rigidly dock within the binding pocket of the receptor and they had shown higher binding capability to the LasR than the native ligand. The study thus indicates that the docked complexes can be considered as stable compounds for further in-vitro experiments.
The clinical isolate Pseudomonas aeruginosa DMC-27b has a genetic profile of a complete QS network with biofilm-forming capacity. Three lead compounds that were proposed in this study will likely be generalizable for other P.aeruginosa strains due to a lack of significant mutation in the LasR protein. But both in-vitro and in-vivo studies are required to check for the potentiality of the suggested QSIs. Based on the findings, this study will continue to provide insights for further research in drug designing and development against clinical isolates of P. aeruginosa as well as other bacteria that use a similar QS system.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)