Keywords
SVs, k-mers, RNASeq, Metagenomics, Mosaic, Long-reads, Hackathon, NGS
This article is included in the Hackathons collection.
SVs, k-mers, RNASeq, Metagenomics, Mosaic, Long-reads, Hackathon, NGS
Structural variants (SVs) are large genomic variations with sizes of at least 50 bps occurring in form of insertions (INS), deletions (DELs), inversions (INVs), duplications (DUPs), and inter-chromosomal translocations.1–3 Recent discoveries have shown SVs to exhibit clinical relevance for multiple diseases beyond classical mendelian diseases, such as multiple types of cancer,4 neurodevelopmental,5 and cardiovascular disorders.6 Nevertheless, detection and evaluation of SVs is still plagued by high false positive and negative rates along with the inaccuracies of breakpoint predictions due to the complex nature of mutations and inherent sample heterogeneities despite the advances in next-generation sequencing technologies.7,8 Third generation sequencing technologies provided by Pacific BioSciences,9 Oxford Nanopore Technologies,10 optical mapping,11 and NanoString,12 as well as new short read technologies provide exciting new tools to enhance SV detection. However, these advancements require new bioinformatic methods for implementation to consequently offer potential for understanding the relationship between these variants and various phenotypes. Accordingly, the objective of the Fifth Baylor College of Medicine & DNAnexus hackathon was to propose and develop novel bioinformatic tools and workflows to improve the use of SV data in disease modeling.
At the Fifth Baylor College of Medicine & DNAnexus hackathon, in August 2023, 49 scientists from 14 nations (see Figure 1) participated in-person and remotely - focusing on different topics on SV related research.
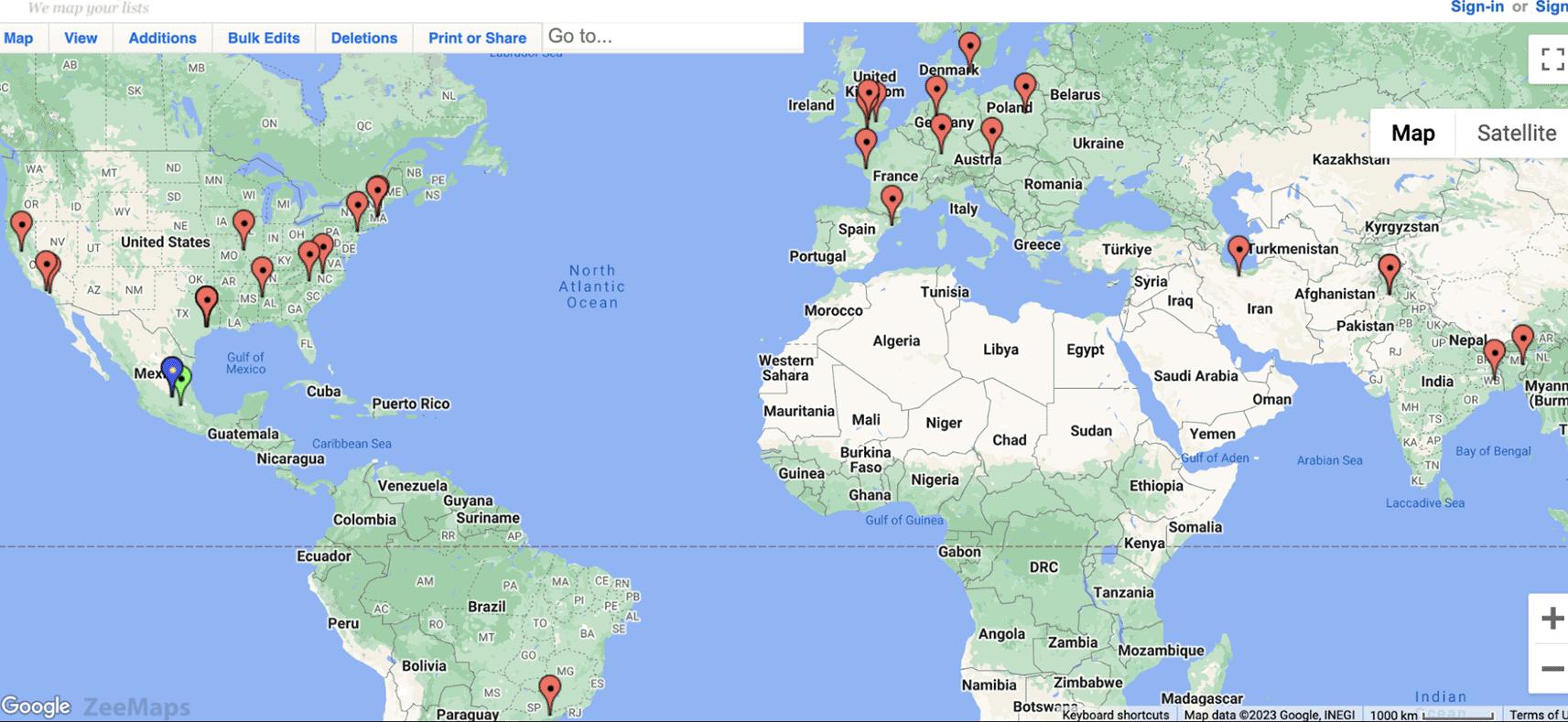
Participants from around the world for the Fifth Annual Baylor College of Medicine & DNAnexus Structural Variation hackathon.
Overall, this manuscript details our tools’ objectives, value-add, implementations, and applications to set the foundation for further concept development. In this article we present six software workflows that were the result of this hackathon.
Alternative splicing is one of the most complicated cellular processes. The joining or skipping of exons from the same gene in various combinations leads to different, but related, mRNA transcripts (isoforms). These mRNA isoforms can be translated to produce diverse proteins with distinct structures and functions. It has been shown that changes in isoform diversity might affect the phenotype, potentially leading to diseases, such as cancer or neurodegenerative disorders. These novel isoforms have been associated with cancer, including novel variants of oncogenes. For instance, novel isoforms and the usage of alternate promoters were found in cell lines in subtypes of gastric cancer.13 Moreover, a positive correlation was found between the presence of particular isoforms of Alzheimer’s disease-associated proteins and pathological severity.14 These findings might help with the development of specific diagnostic biomarkers.
Next Generation Sequencing (NGS), especially transcriptome sequencing (RNA-seq), including short- and long-read sequencing, characterizes gene expression at the isoform level and sheds light on biological processes inside the cell, as well as on how the cell responds to changes in the environment. Short-read sequencing is limited and provides low confidence when predicting alternative splicing, novel exons and junction sites, as well as the characterization of complete isoforms.15,16 Long-read sequencing, however, is expected to overcome the inherent limitations of short-read sequencing. Analysis of long-read sequencing data, that uses multiple variant calling algorithms to detect insertions, deletions, inversions and duplications, found nearly 2.5 times more SVs than short-read data, with ~83% of insertions missing.17 Long-read sequencing technologies, e.g., PacBio High-Fidelity (HiFi) and Oxford Nanopore, have been shown to lower the error rates and to provide high-quality full-length reads that cover the entire length of all isoforms, removing any uncertainty in determining exon composition.
There exist tools that compare sets of short-read transcript annotations, including GffRead (RRID:SCR_018965) and GffCompare,18 AGAT,19 and bedtools20 (RRID:SCR_006646). However, to our knowledge, no tool currently exists which takes advantage of the long-read sequencing data to compare inferred isoforms.
Here we present IsoComp, a bioinformatics pipeline to identify differences in isoform expression between individuals using long-read RNA-seq data. IsoComp can be used to make comprehensive comparisons of isoform profiles between multiple samples, with application in e.g. trio-sequencing or subtyping of cancer cell lines. We have tested IsoComp on HG002 samples (NA27730, NA24385, NA26105, NA24385, NA26105, NA27730) from GIAB (Genome in a Bottle)21 to demonstrate its potential use for distinct isoform detection and comparison.
In the context of individual genome comparison, mutations that appear with very low minor allele frequencies are referred to as rare variants.22 Similarly, mosaicism is observed when unique genetic differences arise at low frequencies within the population of cells in a tissue of an individual giving rise to mosaic variants (MVs).23 Recent studies have shown potential disease implications for certain MVs.23 Since MVs can have low allele frequencies and are mixed in with data from the non-mutated cells in the same sequencing output file, they can be challenging to detect as they may appear as noise in the NGS data. Therefore, several pipelines have been developed or adjusted to extract mosaic single nucleotide, structural or indel variants from whole-genome sequencing data, including Sniffles11 (RRID:SCR_017619), DeepMosaic,24 Mutect2,25 DeepVariant.26 To benchmark and validate the efficiency and accuracy of these methods, we developed two workflows called SpikeVar (Spike in Known Exogenous Variants) and TykeVar (Track in Your Key Endogenous Variants). As input SpikeVar takes data from two different sample sets, and mixes them in a user defined ratio. In contrast, TykeVar generates random mutations in the reference genome that are then introduced into existing sample data at a user defined ratio. Both these methods generate potentially low allele frequency, sporadic events in the resulting dataset.
Species evolve continuously, which causes a drastic shift in the architecture of their genome over time. To consider a single genome as a reference for different studies could produce biases. Bacterial evolution in particular involves different mechanisms like homologous recombinations, horizontal gene transfer (HGT) and mutations.27 Conventional comparative genomic analyses that solely rely on linear reference sequences can introduce biases rooted in reference sequences and potentially disregard the spectrum of population or strain diversity. To mitigate these limitations, pangenomes using a graph genome approach have been proposed, encompassing entire genomes of different strains of species lying under one clade. This approach increases the accuracy of analyses by considering innately the concept that the existence of different haplotypes can cause drastic change in the overall study involving that species.28
Pseudomonas aeruginosa (P. aeruginosa) is a Gram-negative bacterium and an opportunistic pathogen that has been deeply studied due to its significant role in causing serious health concerns in humans. P. aeruginosa presents high genome plasticity, possessing a significant assortment of genes acquired by HGT. These genes are frequently localized within integrons and mobile genetic elements, such as transposons, insertion sequences, genomic islands, phages and plasmids. This genomic diversity results in a non-clonal population structure, and consequently in highly variable strain phenotypes concerning virulence, drug resistance and morbidity. Consequently, this makes P. aeruginosa a prime candidate for a pan-genome graph approach.29,30
Here, we made an amalgamation of several in-silico processes used to create a graph genome of P. aeruginosa, intended for open access within the scientific community. We also compared read alignments methods between a graph-genome and a standard linear genome approach.
Mendelian inconsistencies are identified when a child has a genotype that is not possible given the genotypes of the parents, for example, when a child is homozygous for an allele that does not exist in either parent. Mendelian inconsistency in SV calls can indicate two possibilities: challenges in SV calling leading to false positive or negative calls across the trio, or a genuine de novo SV. De novo SVs are rare, with an estimated rate of 0.16 de novo SVs per genome in healthy individuals.31 Despite their rarity, de novo SVs have been associated with human disease, including Autism Spectrum Disorder, Pulmonary Alveolar Proteinosis and Alzheimer’s disease.32–35 In addition, benchmarking studies have used the rarity of de novo SVs to support the validity of their SV calls under the assumption that calls inconsistent with Mendelian inheritance are likely incorrect.36–38
We present SalsaValentina, a pipeline which identifies putative de novo SVs based on Mendelian inconsistency, and subsequently validates them using a local genome assembly of the region in question. SalsaValentina could assist in diagnosis variants underlying rare diseases, and inform other strategies for more accurate SV calling.
Copy number variation (CNV) is a common form of SV polymorphism where segments of DNA are either duplicated or deleted when compared to a reference genome.39 CNV plays a pivotal role in genome evolution40 and has been associated with phenotypic diversity41 including human diseases.42 To date, the predominant strategies to detect CNVs using whole-genome sequencing data have relied upon analyzing the distribution of mapping coverage across the genome and identifying regions with outlying coverage compared to the background.43 However, these coverage-based approaches are susceptible to ascertainment bias because they can only detect CNV of sequences present in the reference assembly, failing to capture the complete spectrum of CNV within a population. Furthermore, coverage-based CNV detection methods to detect CNV are dependent on high-quality genome assemblies, which are not available for many non-model systems.
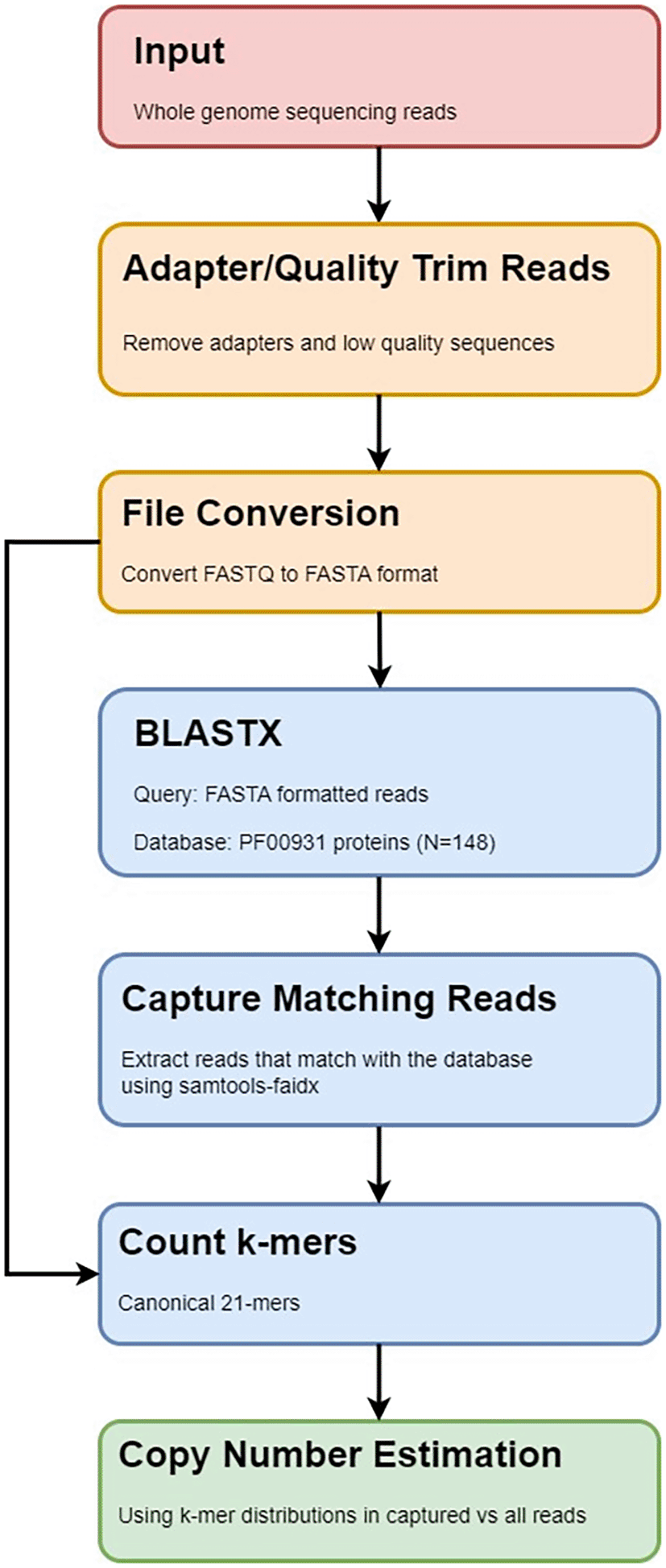
As an alternative approach, we here present PhytoKmerCNV, a tool for estimating copy number of specific sequences using k-mer frequencies derived from whole-genome sequencing reads. Our approach involves comparing the k-mer frequency distributions between reads originating from the sequences of interest to the distribution of frequencies calculated from all sample reads.
Recent studies have shown that SVs are widely present in human genomes. They shape the genome evolution and play important roles in human health and diseases by changing the protein-coding regions, cis-regulatory elements and gene expression profiles.44 While a number of NGS-based SV detection tools have been developed in the past few years, it remains unclear how well these tools perform for the detection of SVs.45
Currently available SV-calling tools roughly fall into three groups depending on the different types of input data used in the SV detection step: mapping-based, assembly-based and mapping-free methods. The mapping-based approaches use SV-related alignment features such as soft-clipped read ends, alignment breakpoints and discordant mates to detect SVs.46 For the assembly-based methods, reads can be assembled directly into contigs (“global assembly”), e.g., in DISCOVAR47 (RRID:SCR_016755) or reads could be aligned to a reference genome first and then reads aligning to each region could be assembled into contigs (“local assembly”), and the contigs (and the corresponding reads) are then aligned to the reference genome for SV calling.48 The mapping-free approach works by checking the genomic signatures (e.g. k-mers) of known SVs directly in the raw NGS reads, as is done in Nebula.49 The alignment-based approach has a number of advantages, including low computing resource requirement and shorter run-time in most cases. By contrast, the assembly-based approach generally requires substantially more computing resources and input sequencing data, but could take advantage of the longer input sequences from the assembled contigs and theoretically, could perform better for SV detection. The mapping-free approach is computationally efficient, but is limited to the genotyping of known SVs and will not be discussed further.
The performance of SV calling tools also depends on the targeted regions and read lengths. While it is generally believed that long read based SV calling tools have better sensitivity, a recent study shows that most large SVs in cancer can be detected without using long reads.50 The targeted regions for SV calling can range from small gene panels to whole exome sequencing to whole-genome sequencing. Targeted panel sequencing and whole exome sequencing are currently dominating in clinical NGS testing due to cost advantages by focusing on protein-coding regions. However, genetic diagnosis by whole exome sequencing can only be made in 25-50% of cases.51 On the other hand, the whole-genome shot-gun sequencing (WGS) data has more uniform coverage and provides a comprehensive view across the coding regions, non-coding regions and intergenic regions. As the sequencing cost continues to drop, whole-genome sequencing is becoming increasingly cost-effective and can serve as a great starting point for detecting SVs and other genetic changes.
The goal of the SV-genie project was to develop a generalized framework to evaluate the performance of SV calling tools for WGS short-read dataset. Specifically, we use the Illumina short-reads data set for GIAB HG002 (ASJ son) as the input, run a number of alignment-based and assembly-based SV calling tools and compare the results with the GIAB HG002 SV dataset to gain insights into the performance difference between mapping-based and assembly-based approaches for the detection of SVs.
IsoComp: Comparing Isoform composition between cohorts using high-quality long-read RNA-seq
The IsoComp pipeline identifies and compares distinct isoforms across multiple samples. Before running the IsoComp pipeline, GTF files need to be created and pre-processed to serve as input to IsoComp. The IsoComp algorithm is outlined in Extended data.
Iso-Seq analysis
The first step is to create GTF files to subsequently serve as input to IsoComp. First, demultiplexed HiFi reads (Q20, single-molecule resolution) from lima (https://github.com/pacificbiosciences/barcoding/) were processed using IsoSeq3 v3.2.2 (https://github.com/PacificBiosciences/IsoSeq). Next, the transcripts were mapped against the GRCh38 (v33 p13) reference genome using Minimap252 (RRID:SCR_018550) (v2.24-r1122; command: minimap2 -t 8 -ax splice:hq -uf --secondary=no -C5 -O6,24 -B4 GRCh38.v33p13.primary_assembly.fa sample.polished.hq.fastq.gz). Then, cDNA_cupcake (v28.0.0) (https://github.com/Magdoll/cDNA_Cupcake) was used to filter away redundant isoforms from the BAM file, followed by filtering low-count isoforms by 10 and discarding 5’-degraded isoforms as they are not biologically significant. Afterwards, SQANTI3 v5.053 was used to generate the final FASTA transcripts and GFF files, as well as the isoform classification reports. The external databases including reference data set of transcription start sites (refTSS), list of polyA motif, tappAS-annotation and Genecode GRCh38 annotation were utilized during the isoform classification by SQUANTI3.53 Finally, IsoAnnotLite (v2.7.3) (https://isoannot.tappas.org/isoannot-lite/) was used to annotate the GTF files obtained from SQUANTI3.53 The workflow, shown in Extended data, outlines each step to generate the GFF files from individual samples of HG002, a necessary pre-processing step for subsequent comparisons using IsoComp.
Isoform clustering and comparison
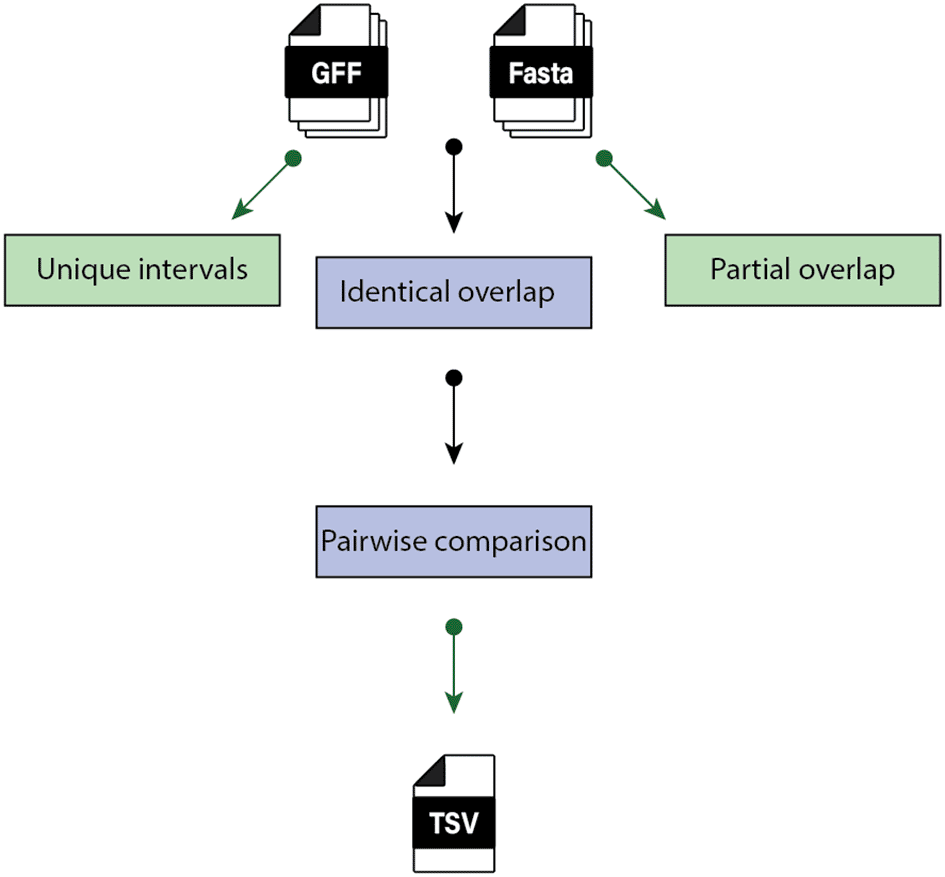
In each GTF file created in the Iso-Seq analysis step, the ‘source’ column is replaced with the base filename (no extension) of the file. Next the GTF files are converted into PyRange objects, which are filtered on the ‘transcript’ feature. Then, overlapping ranges (the start to end coordinates of the transcripts) of clusters are determined. Clusters are sequentially numbered, so that each cluster comprises a discrete group of transcripts with overlapping ranges of coordinates.
In the next step, in each cluster, all isoforms are filtered by the coordinate overlap and compared against one another. If there is only one isoform in the cluster, it is reported as unique. Isoforms with unique or partial overlap of coordinate ranges within clusters are reported as unique, whereas isoforms with an identical overlap undergo a pairwise sequence comparison. The sequence comparison step allows for the detection of variability among isoforms within each cluster.
The workflow of the Isoform clustering and comparison step is presented in Figure 2.
SpikeVar
The SpikeVar workflow outputs a mixed sequencing read dataset in the BAM format, containing reads from one dominant sample and reads from another sample spiked in at a user defined ratio corresponding to the simulated mosaic variant allele frequency (VAF), plus a VCF file annotating the confirmed mosaic variant locations within the mixed dataset (Figure 3(i)). The SpikeVarDatasetCreator takes aligned sequencing reads from sample A and sample B as input. In this step, a spike-in methodology is applied to strategically introduce x% of mutations from one sample to another using samtools54(RRID:SCR_002105) view -s option. Accordingly, sample A is first down-sampled to retain (100-x)% of its original reads, then sample B is down-sampled to x% considering the coverage differences between the samples. Using samtools merge command, both down-sampled datasets are then merged to create a mixed dataset that represents a sequence read dataset with mosaic variants, including SVs, single nucleotide variations (SNVs), and insertions/deletions (indels).
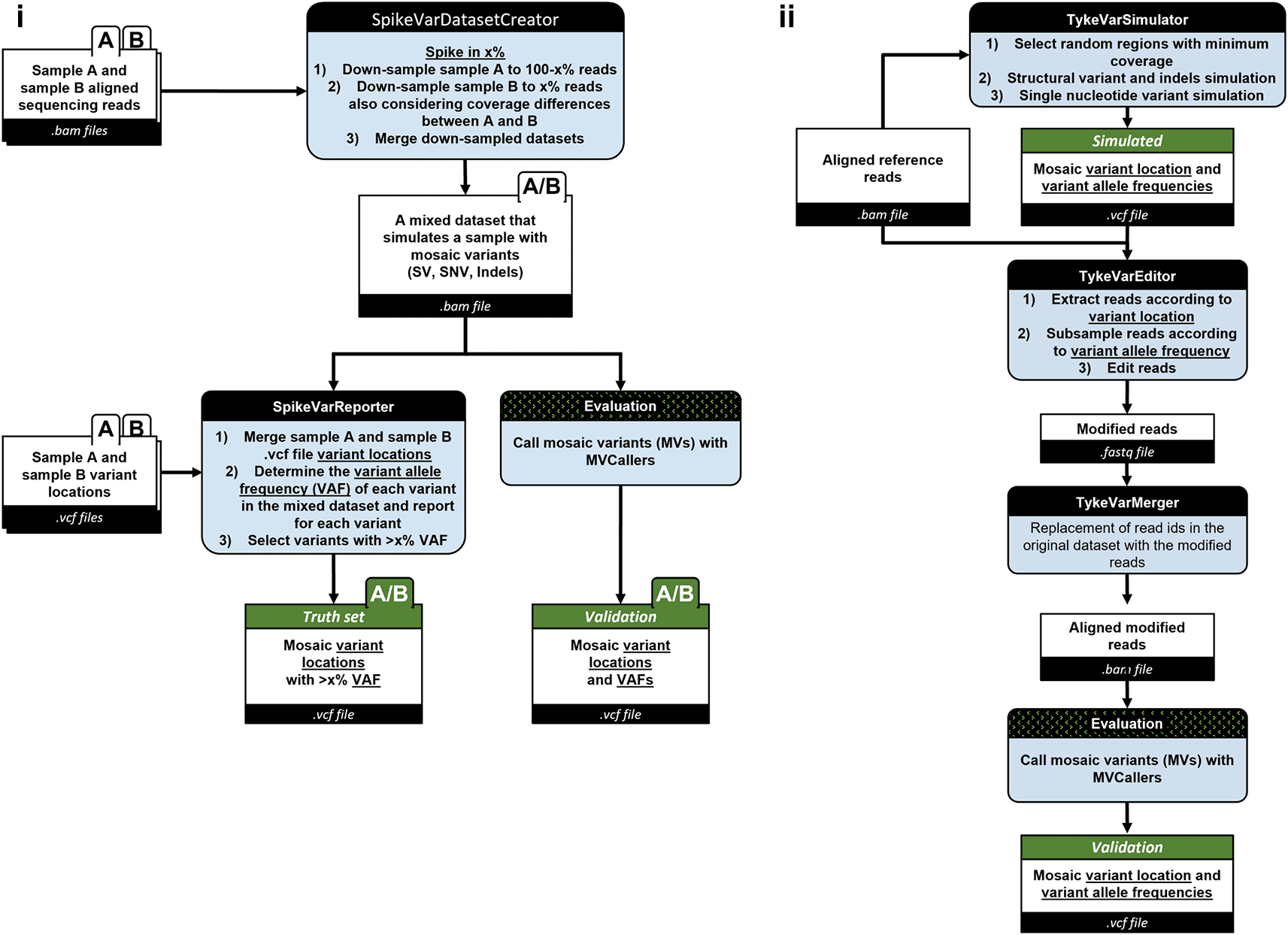
i) SpikeVar workflow and ii) TykeVar workflow, with major steps to assess the sensitivity and accuracy of the mosaic variant callers. (A, B: individual samples, A/B: merged samples, .bam and .vcf: input and output file formats in different steps, Black header boxes: tool or file names, Green header boxes: simulated files or final files used for validation comparisons.)
The SpikeVarReporter then determines VAFs for each variant in the mixed dataset depending on the variant type and sequencing technology SNVs (samtools mpileup command), Sniffles211 (SV and long-reads) (RRID:SCR_017619), and Paragraph55 (SV and short-reads), based on the mixed variant locations derived by merging the VCF files from sample A and sample B using samtools.54 Variants with VAFs exceeding or equal to the introduced mutations (i.e., x%) are then selected to create a truth set for benchmarking using bcftools54 (RRID:SCR_005227).
To assess a mosaic variant caller sensitivity and accuracy, the same mixed dataset is used to call mosaic variants. The output mosaic variant locations and VAFs are then compared to the truth set for validation.
TykeVar
The TykeVar workflow produces a modified aligned sequence file in the BAM format (Figure 3(ii)). This file contains modified reads simulating randomly positioned mosaic variants with user-defined VAF in random locations and is accompanied by a VCF file containing the locations of the simulated mosaic variants with user-defined VAF.
The TykeVar workflow can be broadly split into 3 parts:
1) The TykeVarSimulator takes an aligned BAM file, a reference and several parameters (such as range of VAF, variant sizes). to generate a set of simulated mosaic SVs and SNVs. It does so by choosing a random location and VAF from the given range and then evaluating whether that location has sufficient coverage for the desired VAF. If that condition is met, that variant is added to the output VCF file.
2) The TykeVarEditor is responsible for inserting the simulated variants into the query sequences from the original dataset to generate modified reads with the mosaic variants built-in. The TykeVarEditor accepts a BAM file, reference and the simulated VCF file as input. Then, for each variant, it fetches the overlapping reads from the BAM file, subsamples the reads to get the coverage that satisfies the desired VAF, and traverses the cigar string, query and reference sequences for each alignment to find the exact location to insert the variant. Once a modified read is created, it is written out into a FASTQ file. Note that for all new bases (SNVs or inserts), the q-score of 60 is chosen.
3) The TykeVarMerger re-introduces the modified reads into the original dataset. It does so by first removing the modified read IDs from the input BAM file to create a filtered BAM file. Then, the modified reads are aligned against the reference, and merged with the filtered BAM file. The end result is a BAM file with the same set of read IDs as the original dataset, except for some reads modified to contain the mosaic variants.
The output of this pipeline is thus a modified BAM file and a VCF file which provides the truth set for the mosaic variants.
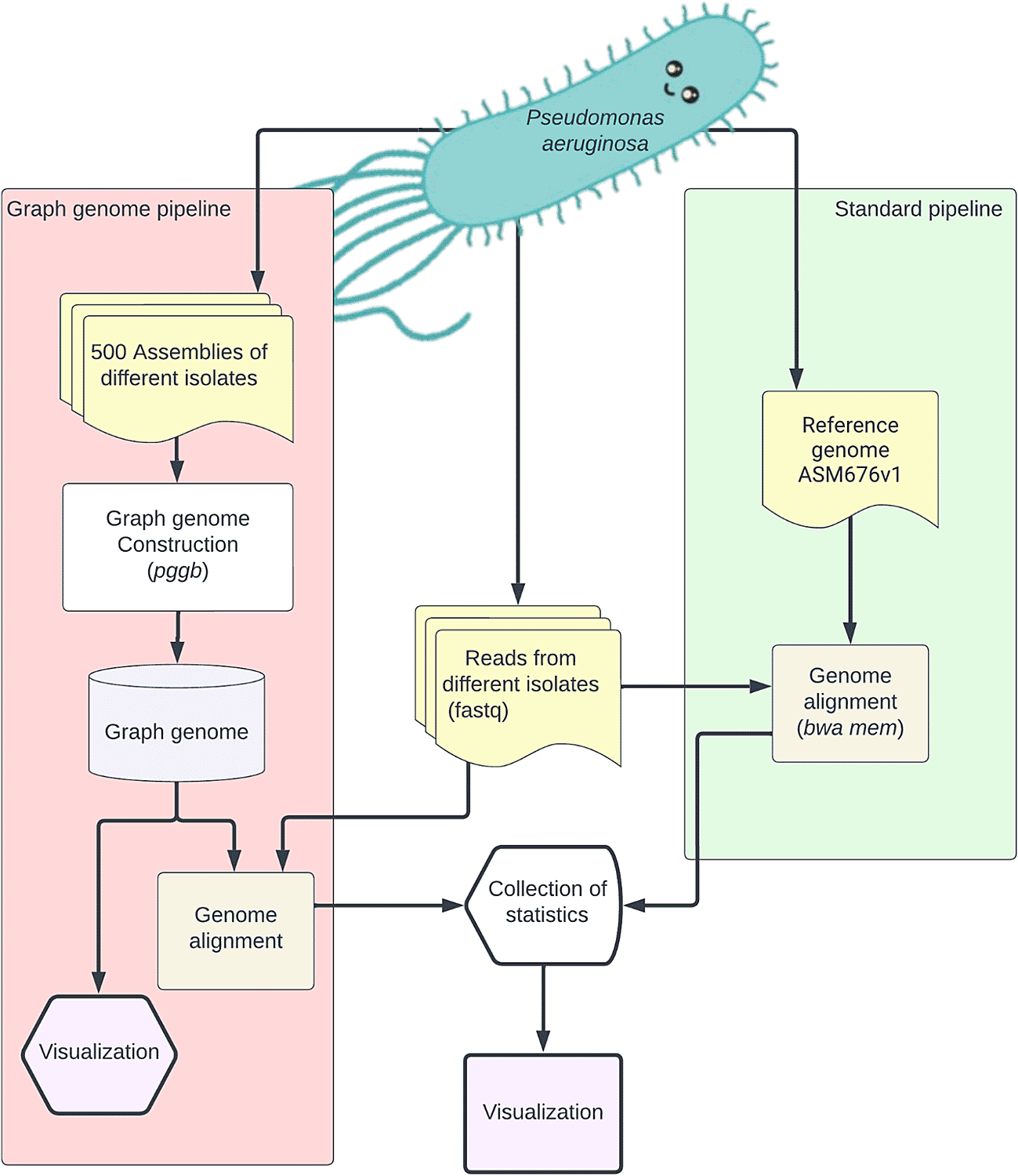
The PGGP process involves two main simultaneous steps (Figure 4). For the pangenome approach, we initially constructed a graph genome by utilizing the accessible assemblies of Pseudomonas aeruginosa. This was accomplished using the pggb tool.56 Then we performed graph alignments with GraphAligner57 to align a dataset of short-read clinical isolates from Sequence Read Archive (SRA)58 (RRID:SCR_004891) to our graph genome. Concomitantly, we downloaded a standard linear reference genome and performed read alignments with the same dataset used for the graph genome using BWA-MEM59 (RRID:SCR_010910). Finally, we compared alignment efficiency between the two approaches. The details of the implementation of PGGP can be found in our Github repository (https://github.com/collaborativebioinformatics/SVHack_metagenomics). They consists of:
1. Graph Approach
a. The graph genome construction: Out of 773 available assemblies, 499 complete P. aeruginosa genomes were downloaded from NCBI. Metadata for the included sequences is included in the linked GitHub repository - https://github.com/collaborativebioinformatics/SVHack_metagenomics/tree/2797b9eec54665258a67ef0277fbd4d06d4e26c7/assemblies_info. Pangenome graphs of varying sizes (5, 10, 20, 50, 100, 500 genomes) were created using the reference NC_002516.2 and complete genome assemblies such that all genomes in the smaller pangenome graphs are contained in the larger graphs. All pangenome graphs were created using the pggb tool (command: pggb -i assembly.fasta.gz -o. -t 16 -n 5 -p 90 -s 5000).56
b. Read mapping to pangenome: Read mapping to single reference Genome: 59 Illumina sequencing datasets including P. aeruginosa (NCBI Taxonomy ID 287) reads were downloaded from the Sequence Read Archive58 (metadata for these samples is included in the linked GitHub repository - https://github.com/collaborativebioinformatics/SVHack_metagenomics/tree/2797b9eec54665258a67ef0277fbd4d06d4e26c7/reads_info). The reference sequence NC_002516.2 (Genome assembly ASM676v1) derived from the PA01 strain was also downloaded. Similar to the single reference genome, reads were mapped to pangenome graphs using GraphAligner (v1.0.10)57 from the Docker image jmonlong/job-graphaligner:latest. Default values related to seeding and extension were used (options: -seeds-minimizer-count 5 --seeds-minimizer-length 19 --seeds-minimizer-windowsize 30 --seeds-minimizer-chunksize 100 -b 5 -B 10 -C 10000).
c. Statistics related to alignment were extracted from the resulting GAM files using vg (1.23.0)60 (RRID:SCR_024369) from the Docker image biocontainers/vg.
2. Linear Genome Approach
a. Reference genome: The reference sequence NC_002516.2 (Genome assembly ASM676v1) derived from the PA01 strain was downloaded and indexed using bwa index ref.fa.
b. Linear read alignment: An array of different genome assemblies of Pseudomonas aeruginosa were downloaded from the NCBI repository. This included the full-length genome of diverse strains isolated from different environments. A total of 59 Illumina sequencing datasets for P. aeruginosa (NCBI Taxonomy ID 287) that were retrieved from the Sequence Read Archive (metadata for these samples is included in the linked GitHub repository - https://github.com/collaborativebioinformatics/SVHack_metagenomics/tree/2797b9eec54665258a67ef0277fbd4d06d4e26c7/reads_info). BWA-MEM (BWA-0.7.17 [r1188])59 was used for short-read alignment to the reference genome for all 59 datasets (command: bwa mem ref.fa read1.fq read2.fq > aln-pe.sam). Quality control statistics were obtained from output files using Picard (2.26.11) (http://broadinstitute.github.io/picard) (RRID:SCR_006525).
True de novo SVs are expected to be rare, however, in practice, a high rate of inconsistent SVs will be identified, indicating false positives or negatives due to noise inherent in SV calling and merging. SalsaValentina creates a ‘naive’ de novo SV candidate list, and develops a QC-framework tool that enables users to visualize the alignments in inconsistent SV regions across the trio and create a local assembly of every de novo SV candidate locus to aid in confirmation of the variant as either a de novo SV or an incorrect call.
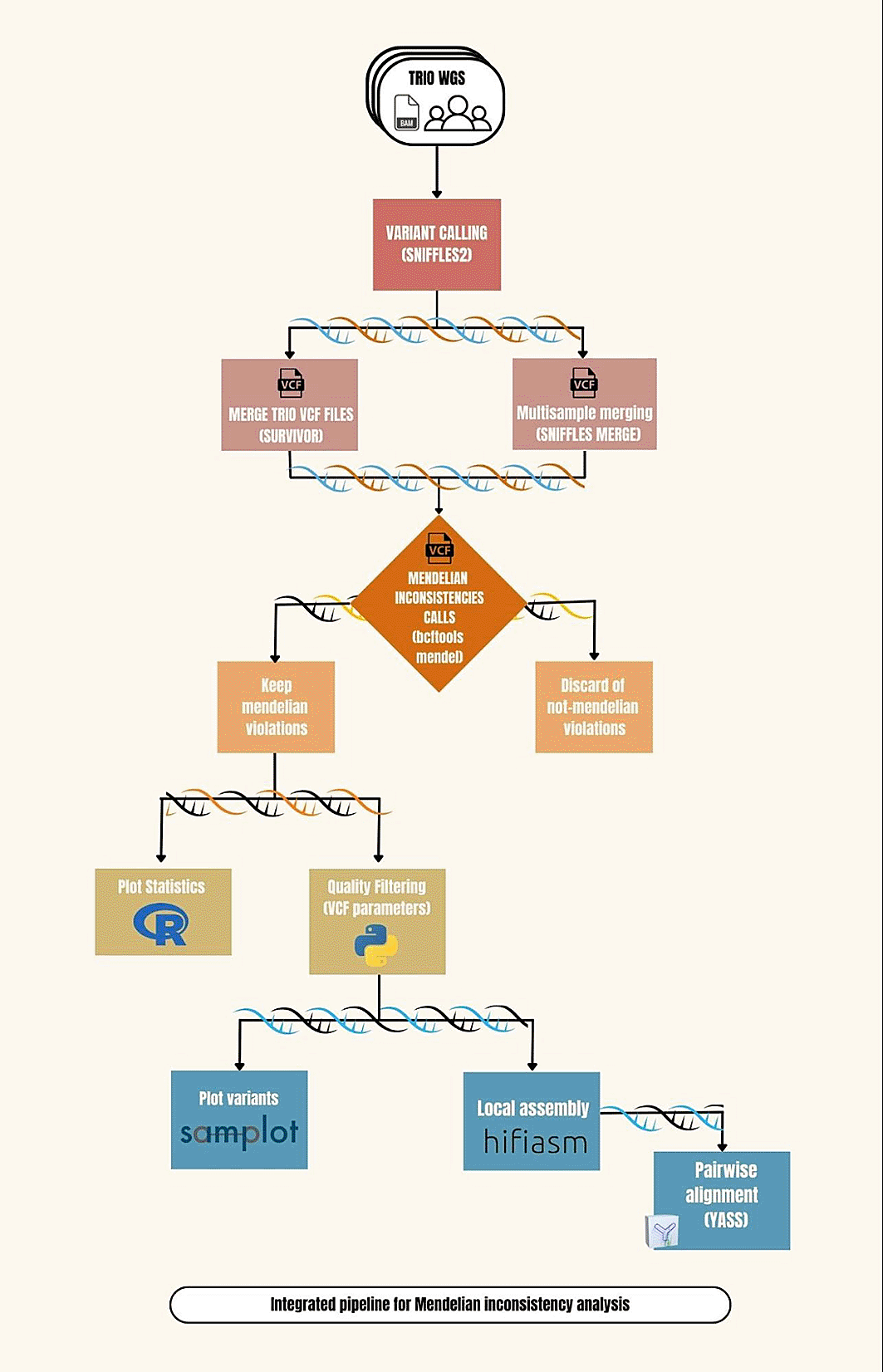
SalsaValentina is an integrated pipeline for Mendelian inconsistency of SVs. We demonstrate the pipeline using the Genome in a Bottle (GIAB) Ashkenazim trio (HG002 son, HG003 father & HG004 mother) sequenced on Sequel II System with 2.0 chemistry and aligned to the GRCh38 genome reference. SVs are called using the Sniffles2 variant caller.11
To merge the SV calls into a single VCF file, two methods are compared: multi-sample SV calling using Sniffles211 and variant merging with SURVIVOR61 (RRID:SCR_022995) using default parameters (https://github.com/fritzsedlazeck/SURVIVOR). Each of the resulting merged VCFs is annotated for Mendelian inconsistencies using the mendelian plugin to bcftools62 ( https://samtools.github.io/bcftools/howtos/plugin.mendelian.html). The positions of each SV inconsistent with Mendelian inheritance is extracted from the merged VCFs and samplot63 is used to visualize the region of each variant in each member of the trio.
To further investigate the validity of the candidate variants, Mendelian inconsistent SVs were filtered to remove breakends (BNDs) and variants involving alternate contigs in GRCh38. The candidates from Sniffles11 multi-sample calling were ranked by coverage. The candidates from SURVIVOR61 merging were filtered for variants with a ratio of variant reads to total read coverage between 0.3 and 0.7, resulting in 48 top candidates. For top ranking candidates, local assembly was performed by extracting reads aligned 50 kb upstream and downstream of the region of interest and assembling with Hifiasm64 (command: --primary; to generate primary and alternate assembly) (RRID:SCR_021069). The YASS65 Genomic Similarity Search Tool web server was used to create dotplots visualizing pairwise alignments of the resulting contigs to GRCh37 to verify the deletion in HG002. SalsaValentina workflow is shown in Figure 5.
PhytoKmerCNV takes raw whole-genome sequencing reads in the FASTQ format as input and produces k-mer distributions for both the total sample, as well as a captured subsample of sequencing reads. The captured reads correspond to sequences of interest in the genome that are captured based on alignment to a protein database. From the k-mer distributions, copy number estimates for the sequences of interest can then be derived from the captured subsample by comparing summary statistics calculated from the respective k-mer distributions for captured versus total reads.
Prior to the pipeline execution, the sequences of interest must be identified, converted to a protein FASTA file, and used to make a BLAST84 protein database. The pipeline then begins with raw sequencing reads in the FASTQ format as input, which can be either uncompressed or compressed with gzip. The sequencing reads are adapter- and quality-trimmed using fastp (0.23.4)66 with the default parameters. The processed FASTQ file is then converted to the FASTA format using seqtk (1.4).67 The sequences are then queried against the protein database using blastx to identify reads putatively originating from the sequences of interest. The BLAST results are filtered to retain hits with a match length≥20 and the E-value<1. Reads with hits to the database are then extracted using samtools faidx. Canonical 21-mers are then counted in the matching reads, as well as the full sample of reads. The sum of k-mer frequencies found in the matching reads, as well as the total sample were then calculated using awk before being fed into an R script that derives copy number estimates from the calculated sums, as well as plotted the k-mer distributions.
As a practical example, we have developed this tool with the goal of estimating the copy number of resistance genes (R genes) for pathogen recognition in a collection of 32 resequenced tomato genomes.68 R genes encode for proteins which recognize pathogen effectors in plants and are classified according to their domain organization, with nucleotide binding site (NBS) and extracellular leucine-rich repeat (LRR) domains.69,70 We selected R genes to test our tool since they evolve rapidly, with copy number variation observed both between and within plant species.71,72 PhytoKmerCNV workflow is shown in Figure 6.
To evaluate the performance of the mapping-based and the assembly-based SV calling methods, we select a number of SV calling tools and analyze Illumina short reads of whole-genome sequencing data to generate SV calls. Once the SV calls are available, the performance of the SV calling protocols are evaluated by comparison with an independently developed high-confidence truth (Figure 7). For this purpose, we used the GIAB HG002 (ASJ son) SV dataset36 as the truth (https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/). The findings could help us to assess the pros and cons for each method and to recommend an optimized SV calling protocol for NGS short reads.
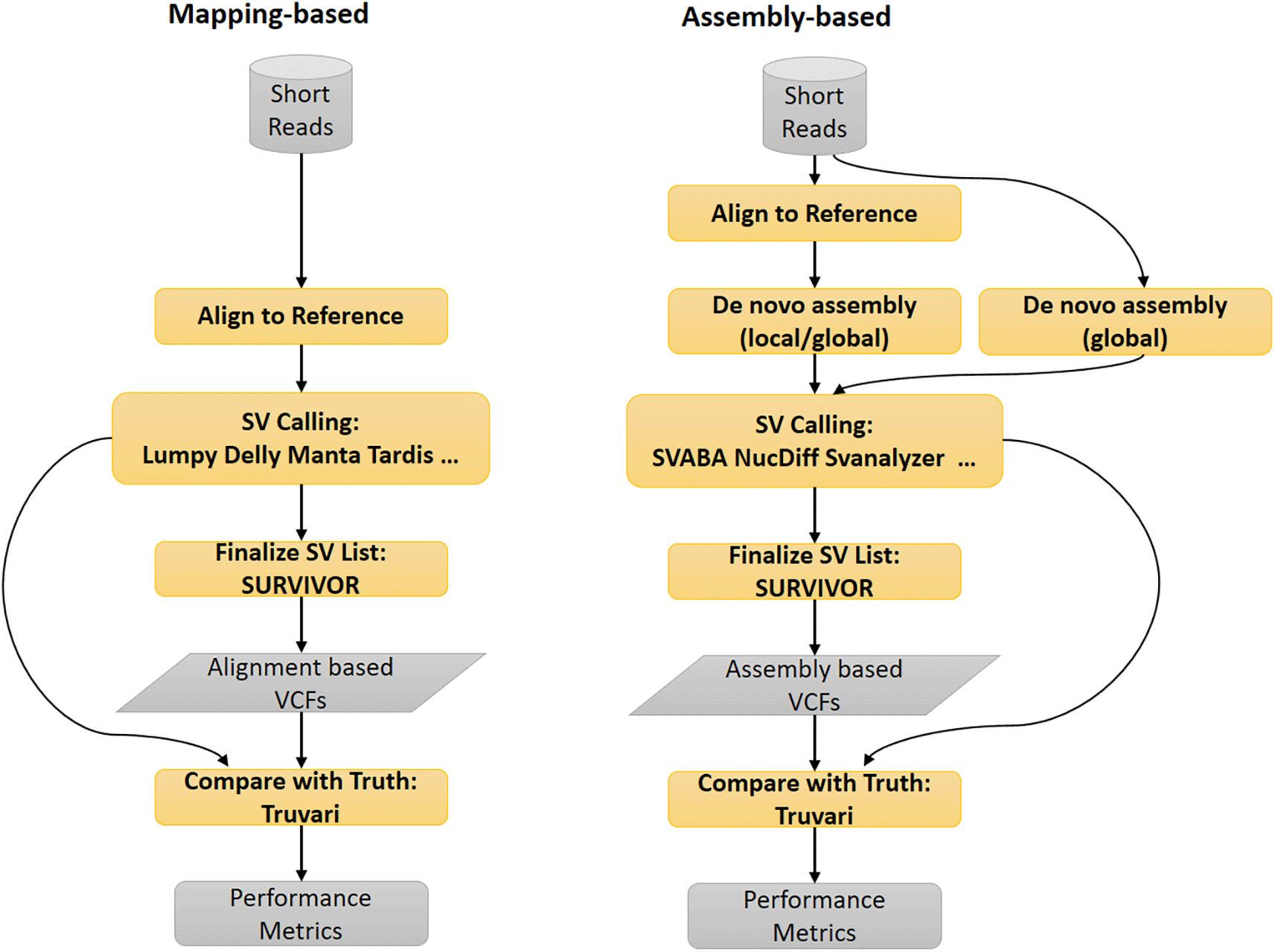
We selected 2x250bp and 300X BAM files (70X coverage) from HG002 (Ashkenazim Trio Son, NA24385, ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/HG002.GRCh38.2x250.bam69ff2c644140bcf2396afed00907e24f; ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_Illumina_2x250bps/novoalign_bams/HG002.GRCh38.2x250.bam; ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_HiSeq_HG002_Homogeneity-10953946/NHGRI_Illumina300X_AJtrio_novoalign_bams/HG002.GRCh38.300x.bam as input for the mapping-based SV calling with Lumpy73 (RRID:SCR_003253), Delly74 (RRID:SCR_004603), Manta75 (RRID:SCR_022997), Breakdancer76 (RRID:SCR_001799), BreakSeq2,77 CNVnator78 (RRID:SCR_010821), Parliament279 (RRID:SCR_019187) and SURVIVOR.61 We also ran dysgu,46 cue80 separately for SV calling. For assembly-based SV calling, the BAM files were converted to FASTQ files and used as input for SVABA48 (RRID:SCR_022998). The individual SV calls and the consolidated SV calls were then compared with the HG002 SV truth dataset to assess the performance by Truvari.81
The recently available Telomere-to-Telomere (T2T) genomes gave us the starting point for developing an alternative ‘self-alignment’ method for evaluating artifacts and false positives from variant callers including small variant detection and SV callers. Since this is a collinear synteny test, we designated this as the Colins Test. Briefly, read alignment was generated by aligning the T2T genome reads back to the corresponding finished T2T reference genome, and the alignment was then used as input for SV calling (see Extended data). SV calls from the SV callers are by definition artifacts and false positives. This alternative performance metric for SV callers serves as an independent complementary approach to the stats generated by Truvari.81 This self-alignment method is also different from SalsaValentina (discussed above), since the self-alignment method does not require trio info to work.
In the Colins Test for GIAB HG002, we mapped GIAB HG002 reads back to the HG002 T2T phased genome assembly (both maternal and paternal haplotypes included) to assess how the Colins Test would perform. We also aligned the HG002 T2T phased genome assembly to the HG19/GRCh37 chr22, and aligned the HG002 T2T maternal to the paternal genome assembly, so then we could inspect the NGS alignments at the matched locations, with the intent of finding the source of errors in the reference based SV calls.
The IsoComp pipeline is written in python and it can be implemented in any Linux-based environment with Python>3.9. After installing IsoComp from PyPI with pip (command: pip install isocomp), the program is ready to use in the command-line terminal. The IsoComp pipeline consists of two steps, run one after the other. The first step (Creating windows) takes GTF files of multiple samples as input and produces a GTF file with clustered isoforms. The second step (Finding unique isoforms) takes the clustered GTF file created in Step 1 as input, as well as a CSV file with information about sources and the FASTA files of particular samples. This step produces a CSV file with unique isoforms. Dependencies include SQANTI3,53 minimap2 (v2.24-r1122),52 samtools (v1.15.1),54 Isoseq3 (v3.2.2) (https://github.com/pacificbiosciences/isoseq/), poetry (v1.6.1), pandas (v1.5.1), biopython (v1.80), pysam (v0.20.0), edlib (v1.3.9), numpy (v1.16), matplotlib (v3.7.1), tqdm (v4.66.1). The code is available inn our GitHub repository: https://github.com/collaborativebioinformatics/isocomp.
The SpikeVar workflow requires Python>3.6.8. The SpikeVar workflow includes two major steps. First, the SpikeVarDatasetCreator takes aligned sequencing reads from two samples to strategically introduce x% of mutations from one sample to another using mosdepth (0.3.2)82 (RRID:SCR_018929) and the two down-sampled datasets are then merged using samtools (1.15.1)54 to create a mixed dataset that represents a sequence read dataset with mosaic variants. In the second step, the SpikeVarReporter determines VAFs for each variant in the mixed dataset using bcftools (1.18)54 based on the mixed variant locations derived by merging the VCF files from sample A and sample B. Variants with VAFs exceeding or equal to the introduced mutations (i.e., x%) are then selected to create a truth set for benchmarking.
The TykeVar package has been tested using Python>3.10. The TykeVar workflow can be broadly split into 3 parts. The TykeVarSimulator takes an aligned BAM file, a reference, and several parameters (such as range of VAF, variant sizes) to generate a set of simulated mosaic SV and SNVs. It does so by choosing a random location and VAF from the given range and then evaluating whether that location has sufficient coverage for the desired VAF. If that condition is met, that variant is added to the output VCF file. The TykeVarEditor is responsible for inserting the simulated variants into the query sequences from the original dataset to generate modified reads with the mosaic variants built-in. For each variant, it fetches the overlapping reads from the BAM file, subsamples the reads to get the coverage that satisfies the desired VAF, and traverses the cigar string, query and reference sequences for each alignment to find the exact location to insert the variant using pysam (0.21.0). Once a modified read is created, it is written out into a FASTQ file. Note that for all new bases (SNVs or inserts), the q-score of 60 is chosen. The parsing and traversing of the VCF, BAM and reference files are performed using APIs from pysam, biopython. SeqIO and NumPy. Lastly, the TykeVarMerger re-introduces the modified reads into the original dataset using minimap2 (v2.24-r1122)52 and bwa-mem2 (v2.2.1).59 Additional non-standard dependencies include NumPy (1.25.2), and BioPython (1.81), all of which are available through the pip package management system.
PGGP is not a workflow that can be run, but an analysis pipeline for a graph genome approach applied to P. aeruginosa. The P. aeruginosa graph genome obtained can also be downloaded from that page, and it is available to the scientific community. This pipeline can be replicated following the instructions in our Github repository (https://github.com/collaborativebioinformatics/SVHack_metagenomics). To re-run this pipeline, we suggest using pggb56 inside a Docker container. Instructions on how to run pggb56 in these conditions can be found in the pggb Github repository (https://github.com/pangenome/pggb). If the user wants to replicate read alignments with GraphAligner,57 even though it was not adequate for short reads (see Results), we recommend running it from inside a Docker container (https://github.com/maickrau/GraphAligner). Other aligners, such as vg Giraffe60 might be more adequate for short reads, however. For a linear genome alignment, we used the reference sequence NC_002516.2 (Genome assembly ASM676v1) as a reference genome, and performed alignment with BWA-MEM (BWA-0.7.17 [r1188]).59 Lastly, vg60 was used to obtain statistics from the graph alignments, while Picard tools (2.26.11) (http://broadinstitute.github.io/picard) was employed to assess the quality and the statistics of the aligned reads aligned to the graph or linear genomes respectively. For more details, see the Github repository (https://github.com/collaborativebioinformatics/SVHack_metagenomics).
SalsaValentina is implemented as a Snakemake pipeline, requiring the sample names and BAM files of the mother, the father, and the child (trio) and a reference genome. The pipeline calls SVs using Sniffles (v2.0.7),11 then merges the variant calls across the trio using both Sniffles11 multi-sample calling and SURVIVOR (v.1.0.7).61 Mendelian inconsistencies are identified with bcftools mendelian plugin (v1.17)62 and automatically visualized with samplot (v1.3.0).63 The pipeline outputs merged VCF files, a text file of Mendelian inconsistencies, and a PDF file with visualizations from samplot.63 Additional scripts, dependent on the Python version (v3.9) and the Pandas module (v2.1.3), enable filtering of the de novo SVs identified by either Sniffles11 or SURVIVOR61 and local assembly of selected regions of interest using samtools to extract regions from the BAM file and hifiasm (v0.19.6).64
The main PhytoKmerCNV pipeline is provided as a Bash script that can be deployed on any Unix system, although the pipeline was initially built and executed on AWS via DNAnexus. Software dependencies can be installed via conda/bioconda using the provided YAML file and include Python≥3.10.12, R≥4.3.2, jellyfish (2.3.0)83 (RRID:SCR_005491), ncbi-blast+ (2.14.1),84 seqtk (1.4),67 sra-tools (3.0.7) (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software), samtools (1.17),54 fastp (0.23.4),66 and awk. Further dependencies include the following R libraries: pacman, ggplot2, cowplot, and ggpubr.
The SV-Genie pipeline has two major branches executed via shell scripts. The first branch is for the mapping-based SV calling, which uses BAM files as input for SV calling with Parliament2 (v0.1.11),79 executing SV callers such as Lumpy (v0.2.13),73 Delly (v0.7.2),74 Manta (v1.4.0),75 Breakdancer (v1.4.3),76 CNVnator (v0.3.3),78 and followed by SURVIVOR (v.1.0.7).61 This branch also runs dysgu (v1.6.0)46 and cue (v0.2.2)80 for SV-calling. The second branch is for the assembly-based SV calling, where the BAM files are converted to FASTQ files and used as input for the assembly-based SV calling via SVABA (v1.1.3).48 The individual SV calls and the consolidated SV calls are then compared with the SV truth dataset to assess the performance by Truvari (4.1.0).81 To evaluate the impact of different read coverage on SV calling, we used seqtk (1.4)67 or samtools (1.17)54 to generate decimated bam files as input for SV calling.
In our approach, we aim to compare transcripts based on their composition rather than solely relying on coordinates, distinguishing our method from others. We have developed a tool called Isocomp, publicly available on GitHub at “https://github.com/collaborativebioinformatics/isocomp” under the MIT License. IsoComp can be installed using pip (command: pip install isocomp==0.3.0) and comprises two main steps. The initial step involves creating comparison windows (command: isocomp create_windows), which takes a GTF file for the samples to be compared and a transcript file as input, producing a cluster file for all samples that serves as a seed for the subsequent step. The next step utilizes the IsoComp algorithm (command: isocomp find_unique_isoforms) and the output from the previous step to compare shared transcripts between samples based on the composition. This step outputs unique transcripts that may overlap with other transcripts in the compared samples but differ in sequence composition.
We applied our tool to two publicly available samples, namely NA24385 and NA26105, and successfully differentiated isoforms between those two samples. However, further development is required.
The refinement and clustering steps generated in the Iso-Seq analysis step full-length HQ transcripts of high quality (i.e., predicted accuracy≥Q20) are shown in Table 1, Table 2 and Table 3
| Samples | Total number of reads | Alignment (%) | Mapped reads | Unmapped reads |
|---|---|---|---|---|
| NA26105 | MM2 | 99.04 | 203607 | 1983 |
| NA27730 | MM2 | 99.35 | 204553 | 1331 |
| NA24385 | MM2 | 99.65 | 409905 | 1444 |
| Sample | Number of genes | Number of transcripts | Number of exons |
|---|---|---|---|
| NA26105 | 5433 | 7632 | 72940 |
| NA27730 | 5459 | 7765 | 74906 |
| NA24385/HG002 | 5656 | 9710 | 51196 |
Table 2 shows the basic statistics of the alignment of each sample (HG002: NA27730, NA24385, NA26105) to the reference genome.
Table 3 shows the isoform classification report generated by SQUANTI3.
The output results (supplementary table 4) were performed by running IsoComp on DNAnexus (total running time: 15 mins, with 16 CPU and 8 GB RAM). Our tool can find and compare intervals, multithreading, easy to install and generate a convenient TSV output file.
In Figure 8, we represent the cluster sizes, which indicate the number of transcripts whose intervals overlap and how many clusters contain such transcripts. We obtained this data by comparing three replicates of the HG002 sample, which is publicly available in GIAB (ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore/guppy-V3.2.4_2020-01-22/HG002_hs37d5_ONT-UL_GIAB_20200122.phased.bam).

Using the SpikeVar workflow, we successfully spiked in the Genome in a Bottle (GIAB) sample HG002 (ASJ son)36 (https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/) at a 5% concentration into sample HG0733 (Puerto Rican female) (ftp://ftp.sra.ebi.ac.uk/vol1/run/ERR398/ERR3988823/HG00733.final.cram), to result in a 5% mosaic variant allele frequency (VAF). Figure 9(i) displays the successful detection of a deletion of 287 bp on chromosome 5 at a 5% rate. This deletion originated from the HG002 sample and was not present in the original HG00733. Similarly, Figure 9(ii) shows a 341 bp long insertion at a 5% VAF originating from HG002.
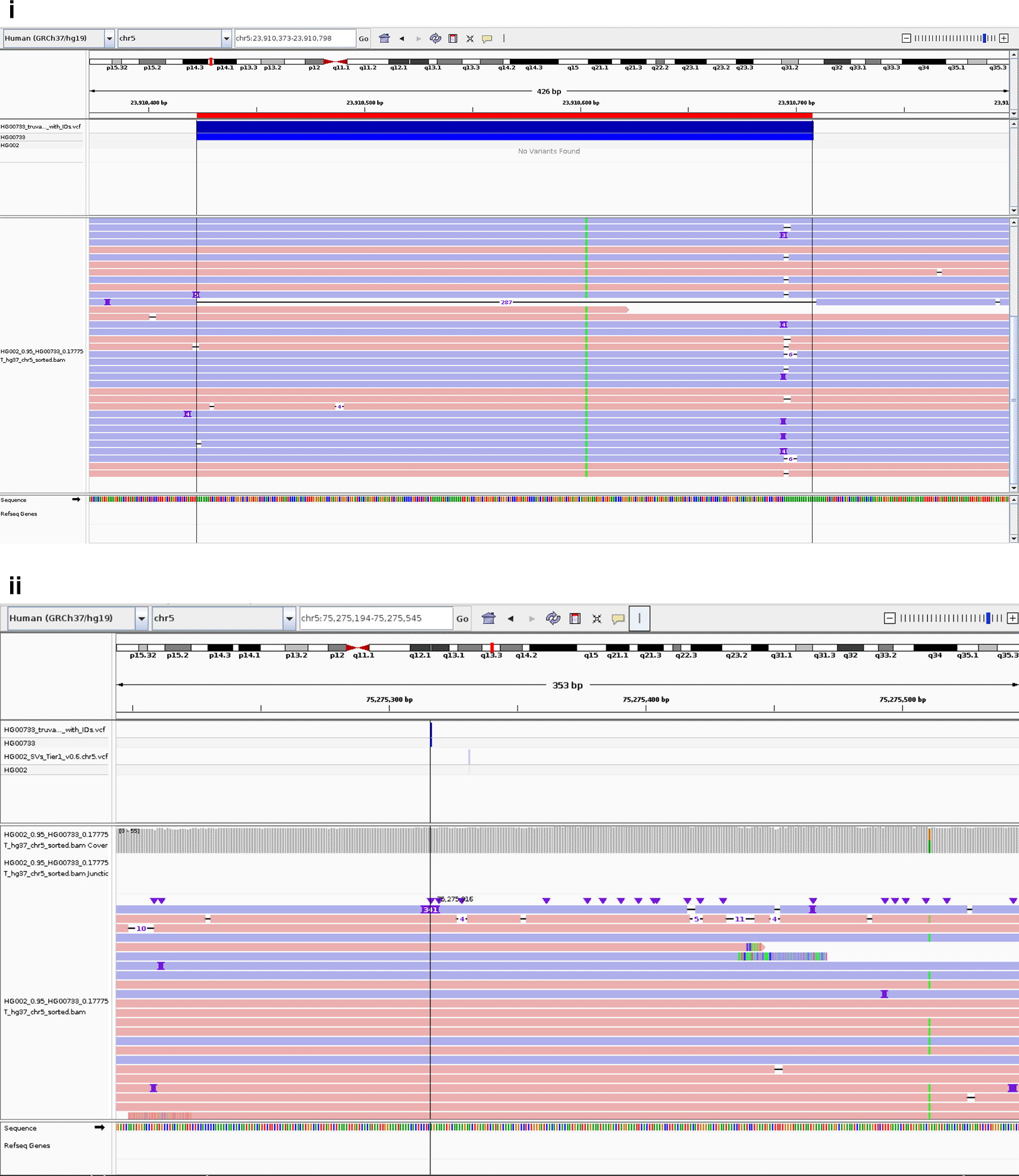
Screenshot for SVs (i) deletion and (ii) insertion from HG002 and was successfully spiked in the HG00733 BAM.
We successfully used the TykeVar workflow to modify reads of HG002 directly at their reference position by including artificial mutations. To demonstrate the wide application of our tool, we generated a random distribution of allele frequencies between 1% and 40%, which can be seen in Figures 10(i) and (ii). Figure 10(i) shows the simulation of a 5952-bp long insertion which was simulated at 22% VAF, while Figure 10(ii) shows the simulation of a A>T mutation on chromosome 22 at 8% VAF.
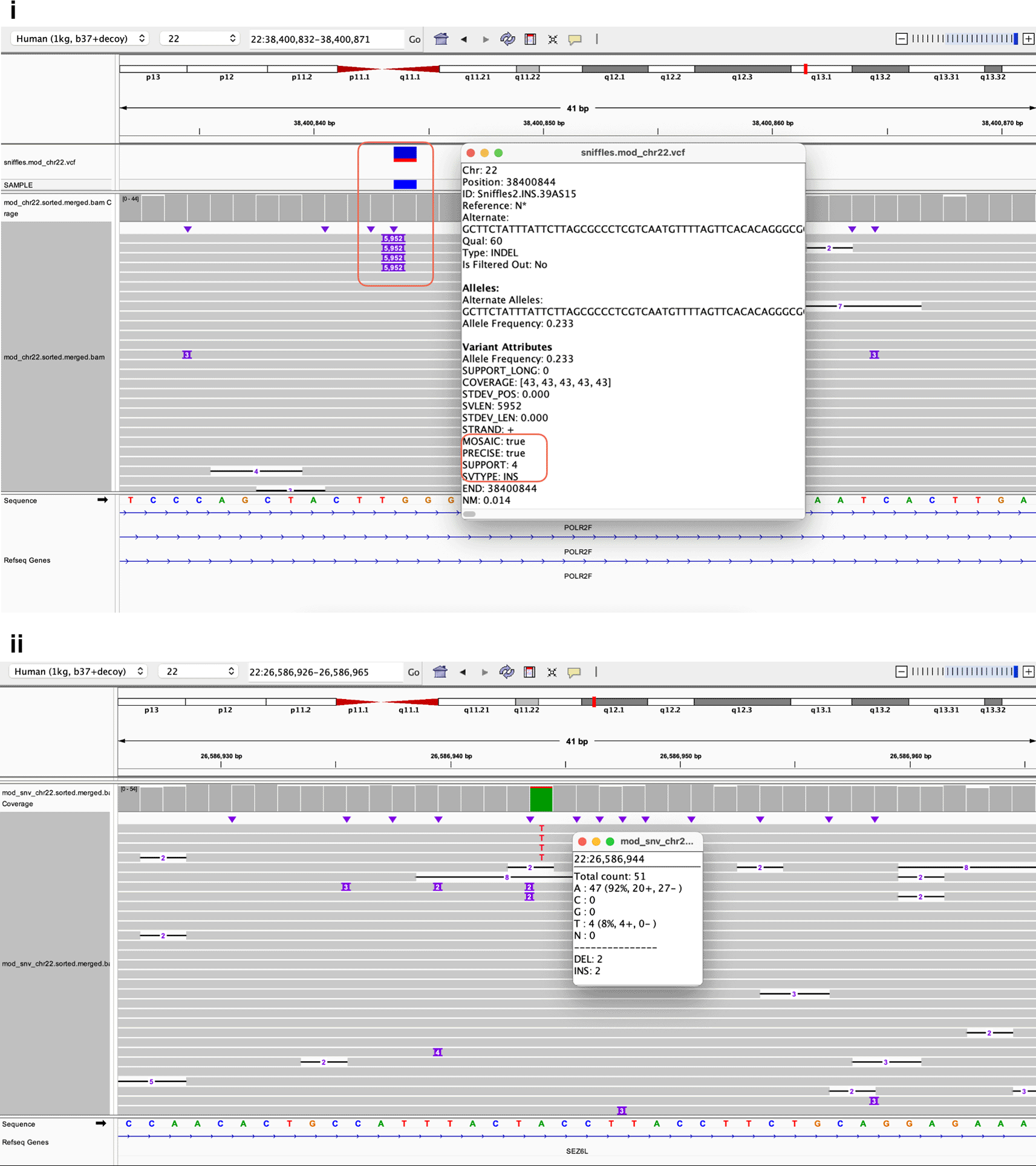
(i). A mosaic insertion introduced into the reads by modifying a subset of the reads. ii). A mosaic SNP introduced into the reads by modifying a subset of the reads.
The simulated data generated by both SpikeVar and TykeVar workflows include simulated SNVs, SV, and indels and is therefore optimal for the comparison and benchmarking of different mosaic variant callers. Mosaic variant caller sensitivity and accuracy can be determined for variable VAFs and read coverages to determine minimum requirements for detection. To validate long SVs and indels detection, SpikeVar is a more suitable workflow for the creation of a validation dataset as it uses naturally occurring variants for spike-in and is not restricted by the read length. Moreover, in simulated datasets created by the TykeVar workflow, haplotypes remain unchanged and therefore TykeVar datasets are more suitable for phasing dependent callers. Both SpikeVar and TykeVar can be applied to long- and short-read whol-genome sequencing files. Hence, different technologies (ONT, Pacbio, Illumina) can be assessed for their suitability of mosaic variant detection.
For the graph genome construction, we built graph representations for Pseudomonas aeruginosa using 5, 10, 20, 50, 100, and 500 genomes (see Extended data). As more assemblies are used, the graph grows in complexity and more regions of the genome are accessory regions and not core. Observing the sequenced alignments of the 20 assemblies used to create that graph, one can see big chunks of the sequences being absent from most of the different isolates, while others are present in all. The assembly accession numbers with additional details can be found in the metadata table in our GitHub repository: https://github.com/collaborativebioinformatics/SVHack_metagenomics.
Linear Read Alignment
As part of the PGGP project, we wanted to compare our graph approach to the regular linear genome alignment approach. We obtained 59 Illumina read datasets from SRA and aligned them to the ASM676v1 reference genome (see Methods). We can observe the alignment efficiency for this approach (see Extended data). To gauge graph alignment accuracy, we also developed a method for graph based MAPQ sampling accuracy check. The quality scores provided in the GAM files are tricky to interpret as we have few details about the calculation method used here and it might be different from linear alignments MAPQ scores calculation methods.57 To count the number of aligned reads on the graph, a criteria for a valid aligned read needs to be defined. Here we look at the distribution of the quality scores found for one of our GAM files to get an idea of its range and have a better overall insight of the threshold. This helps to choose an appropriate threshold to filter the aligned reads and count the one that passes (see Extended data). Based on the distribution of the score, we plotted the selectivity of the filter based on the value of the chosen threshold (see Extended data).
Finally, a comparative analysis between the aligned reads between the pangenome and a single reference genome for different assemblies was done, which shows a better view of the difference in the analysis using a single reference genome and a pangenome (Figure 11).
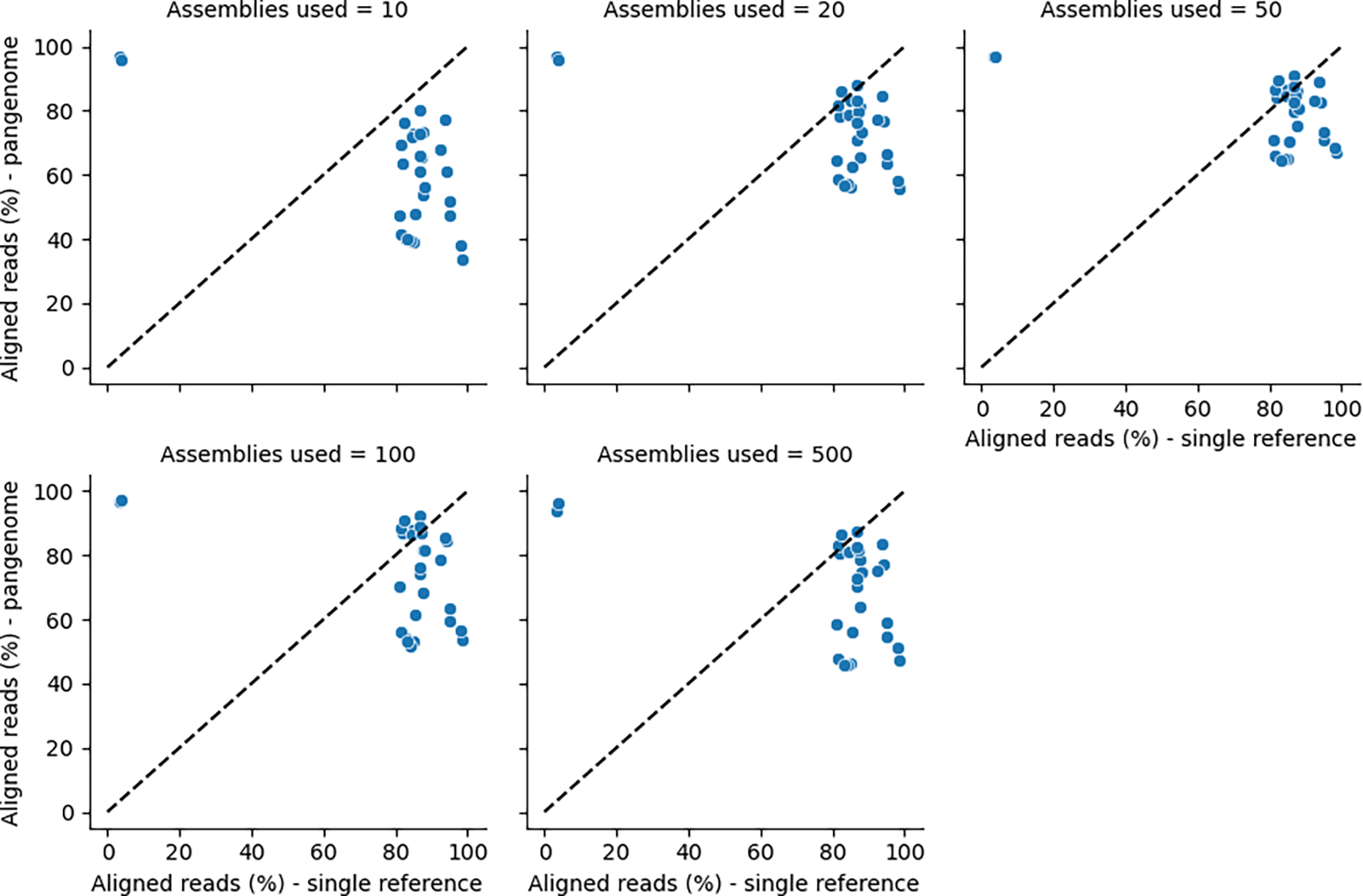
A comparison between the aligned reads between the pangenome and a single reference genome for different assemblies.
Our pipeline offers a valuable resource for the scientific community: a collection of graph genomes using different numbers of isolates, representing different iterations of the Pseudomonas aeruginosa pangenome. These graphs were constructed using a carefully curated list of publicly available genome assemblies from bacterial isolates. The list is also provided alongside the graph genomes, allowing for transparency and reproducibility. Additionally, the pipeline provides a flexible framework for generating various iterations of these graph genomes. This enables users to experiment with different parameters and isolate selections, further tailoring the resource to their specific research needs.
SalsaValentina compares two different methods of merging SV calls within the trio: multi-sample calling using Sniffles11 and merging using SURVIVOR.61 The two methods give different numbers of overall SV calls within the trio, as well as percentages of SVs that are inconsistent with Medelian inheritance. We found a total of approximately 32,000 SV calls in our merged call set using either the Sniffles11 multi-sample calling or SURVIVOR.61 For the Sniffles11 multi-sample calling, 5.2% of these were Mendelian inconsistent, while for SURVIVOR61 2.4% were inconsistent (Extended data). The different number of inconsistent SV calls between the two methods is due to differences in genotype assignment between the tools, with SURVIVOR61 treating some variants as missing, whereas Sniffles11 reports them as reference.
A Mendelian inconsistent deletion was identified in HG002 at chr7:142,786,222-142,796,849 by the Sniffles11 multi-sample calling method (Figure 12(A)). This is in the T-cell receptor beta locus and thus, likely the result of somatic recombination rather than a de novo germline variant. However, it can still be used to demonstrate the usability of our method. This deletion was called heterozygous with 12 reads supporting the reference and 13 supporting the variant in HG002, while it was homozygous reference supported by 45 and 44 reads respectively in HG003 and HG004. In addition, GIAB previously reported a de novo deletion in HG002 at chr17:51,417,826–51,417,932 using GRCH37 reference as part of their v0.6 SV benchmark set, which was derived from high confidence calls supported by multiple methods.36 This deletion was also identified in this study, at chr17:53,340,465-53,340,571 when using GRCH38 as reference (Figure 12(B)). This heterozygous deletion was supported by 30 reads and the reference at this location by 27 reads, while the parents had only reads supporting the reference allele (65 in HG003 and 72 in HG004).
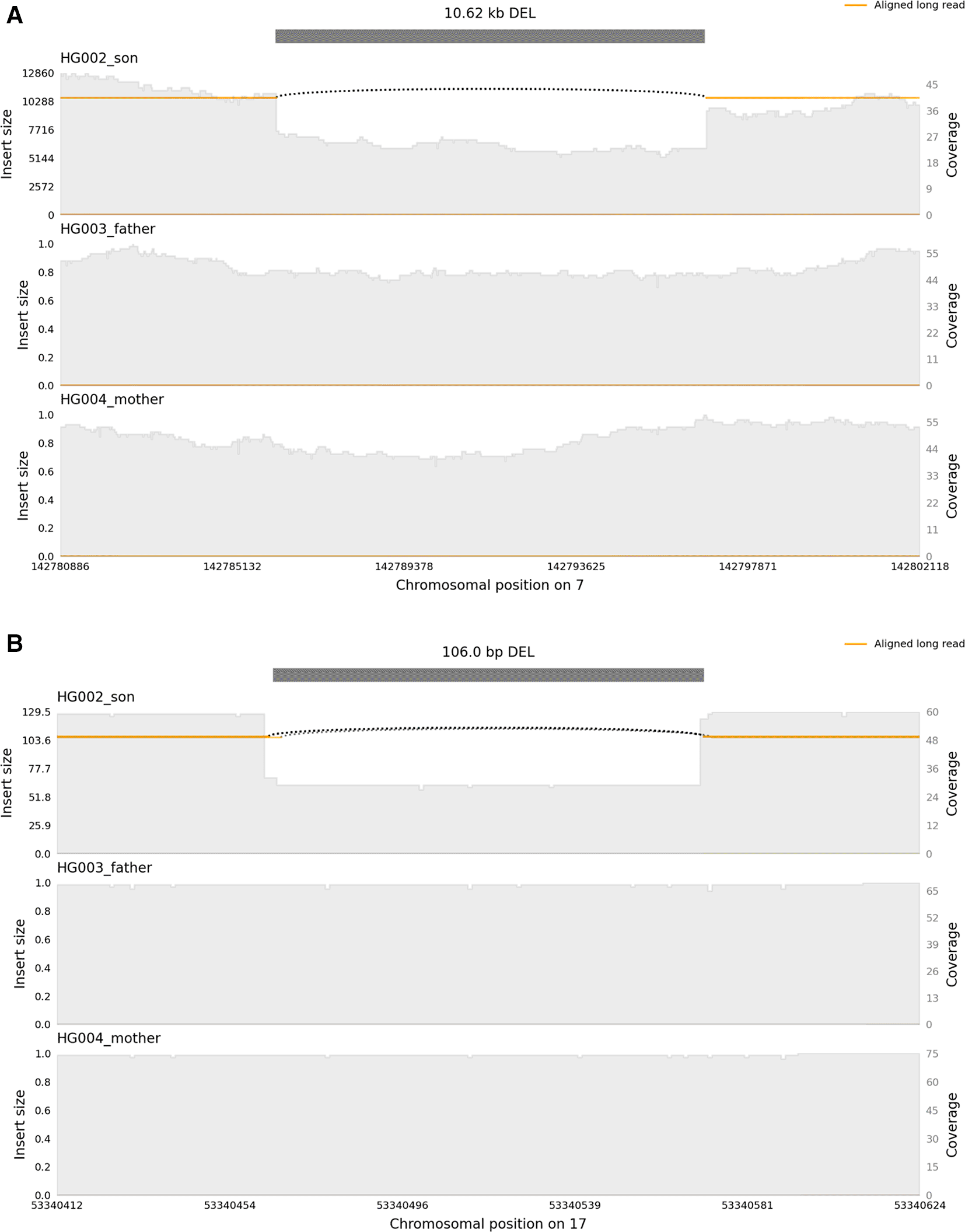
Candidate de novo deletions at (A) chr7:142,757,892-142,824,789 and (B) chr17:51,417,826–51,417,932. The top panel shows a deletion in HG002, which is absent in the parents (father HG003 middle panel, and mother HG004 bottom panel).
SalsaValentina can aid users in identifying and confirming SVs that demonstrate Mendelian inconsistency. We envision two primary use cases. First, genuine de novo SVs may be candidates for rare disease diagnosis. Second, candidate SVs that are mendelian inconsistent due to coverage issues or variant calling inconsistencies help to inform the error modes of sequencing and software for SV calling, to refine methods for accurate SV calling.
We executed the PhytoKmerCNV pipeline on a dataset of 32 resequenced tomato genomes and compared the CNV estimates produced by the pipeline with empirical NBS-LRR genes parsed from genome annotations. Briefly, we counted the number of genes with the NBS-LRR domains among annotated peptides for each genome and used the resulting value as a relative ground truth value, upon which the k-mer estimates were compared to determine the accuracy of our gene copy number predictions. The corresponding results are shown in Figure 13. Our approach did not yield a significant result, thus we were unable to confidently infer the copy numbers for each NBS-LRR gene in the resequenced tomato genome. There is much room for further refinement of PhytoKmerCNV.
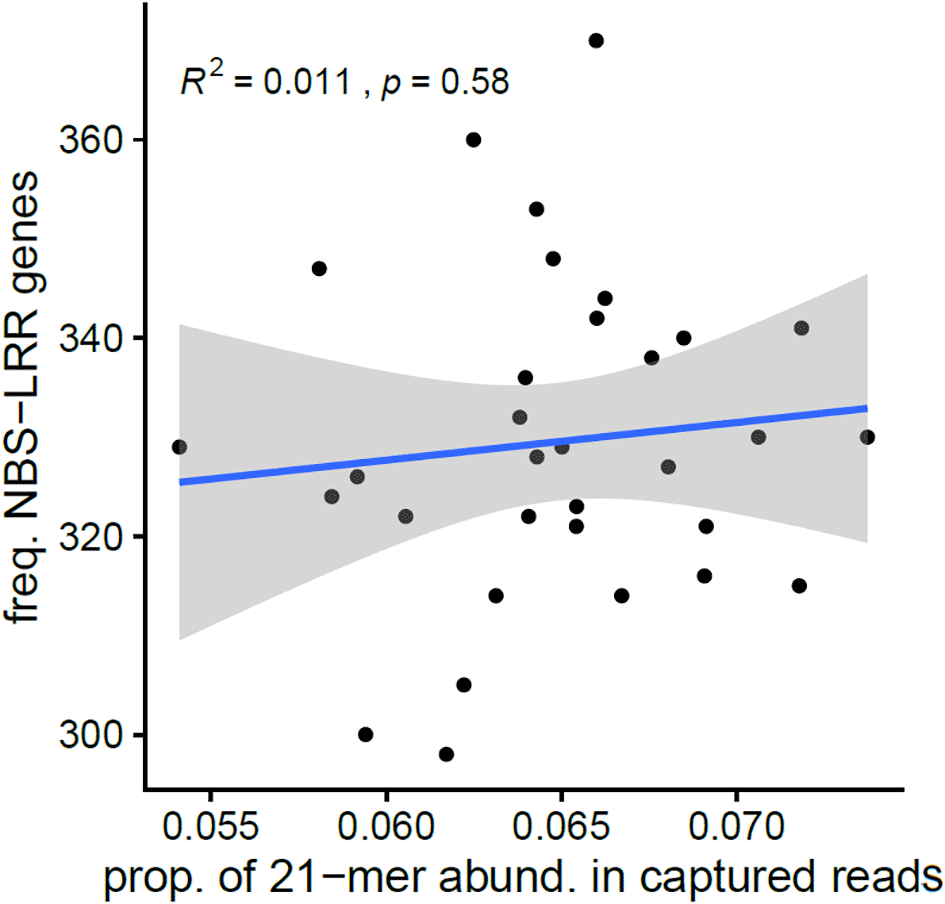
The regression analysis shows the relationship between the number of NBS-LRR genes estimated from the gene annotations compared to the 21-mer based abundance estimates derived from captured reads. The R2 value is 0.011 indicating a very low correlation between the above variables, while the p-value of 0.58 suggests that the results are not statistically significant.
There are three potential use cases of PhytoKmerCNV. First, it would be interesting to compare CNV patterns of the NBS-LRR genes within a species and/or across multiple plant species, highlighting variations and conserved patterns. Second, it would be informative to be able to estimate CNV in a non-model plant genome lacking extensive resequencing data, especially including gene identifiers and inferred copy numbers. Third, extending this approach to assess CNV using low-pass sequencing data would reduce associated costs. These practical use cases illustrate the versatility of PhytoKmerCNV for CNV analysis in plant genomes. Researchers can adapt the tool to address a wide range of research questions, from investigating genetic diversity to understanding the functional implications of CNVs in plant biology.
We ran the SV-Genie pipeline on the GIAB HG002 2x250bp WGS Illumina short-reads as a use case. Specifically, we executed the mapping-based SV-calling including Parliament2,79 dysgu46 and cue,80 as well as the assembly-based SV-calling SVABA.48 We then compared the SV calls with the GIAB HG002 SV reference data set v0.636 via Truvari.81 The performance stats are summarized in Extended data.
This use case gave us a number of insights:
1. SV callers have false negative rate of 50-60% or higher and false positive rate is also high, consistent with previous observations.46,79
2. SV callers have much better performance for deletions than for duplications/insertions, suggesting more challenges for duplication/insertion SV calling.
3. Parliament279 is designed to launch all six included SV callers, but two of these SV callers (breakdancer76 and lumpy73) failed to run, even though all these SV callers were included as part of the Docker image.
4. Parliament279 generated final SV calls from the four successful SV caller runs (breakseq2,77 cnvnator,78 delly2,74 and manta75) even when two of the included SV caller failed to generate any results.
5. The dysgu46 SV caller alone out-performed Parliament279 with better recall and F1-score, even though Parliament279 integrated the results from multiple SV callers.
6. The SV calling performance has a strong dependency on the coverage, where the 70X coverage for 2×250bp data has the best performance. However, the 300X coverage dataset has significantly worse performance, suggesting the possibility of an optimal coverage for SV calling, or the read length difference between the 300X data and the 2×250bp data.
7. SVABA48 is the only assembly-based SV caller evaluated, but its performance with default settings is far worse than the mapping-based SV callers. SVABA48 for 2×250bp data (70X) completed in a few hours by just assembling clipped/discordant/unmapped/gapped reads (default). When the -r all option is turned on to generate assembly for the whole genome, the job ran for seven days without completion on a large DNAnexus instance (mem2_ssd1_v2_x96: 375 GB total memory, 3348 GB total storage, 96 cores).
To complement the performance metrics generated by Truvari,81 we created a genome browser instance containing the read alignment with T2T reference genome to demonstrate manual review and confirmation. The screenshots shown in Figure 14 and Extended data were created with an automated script that prepared the alignments and the genome browser setup.
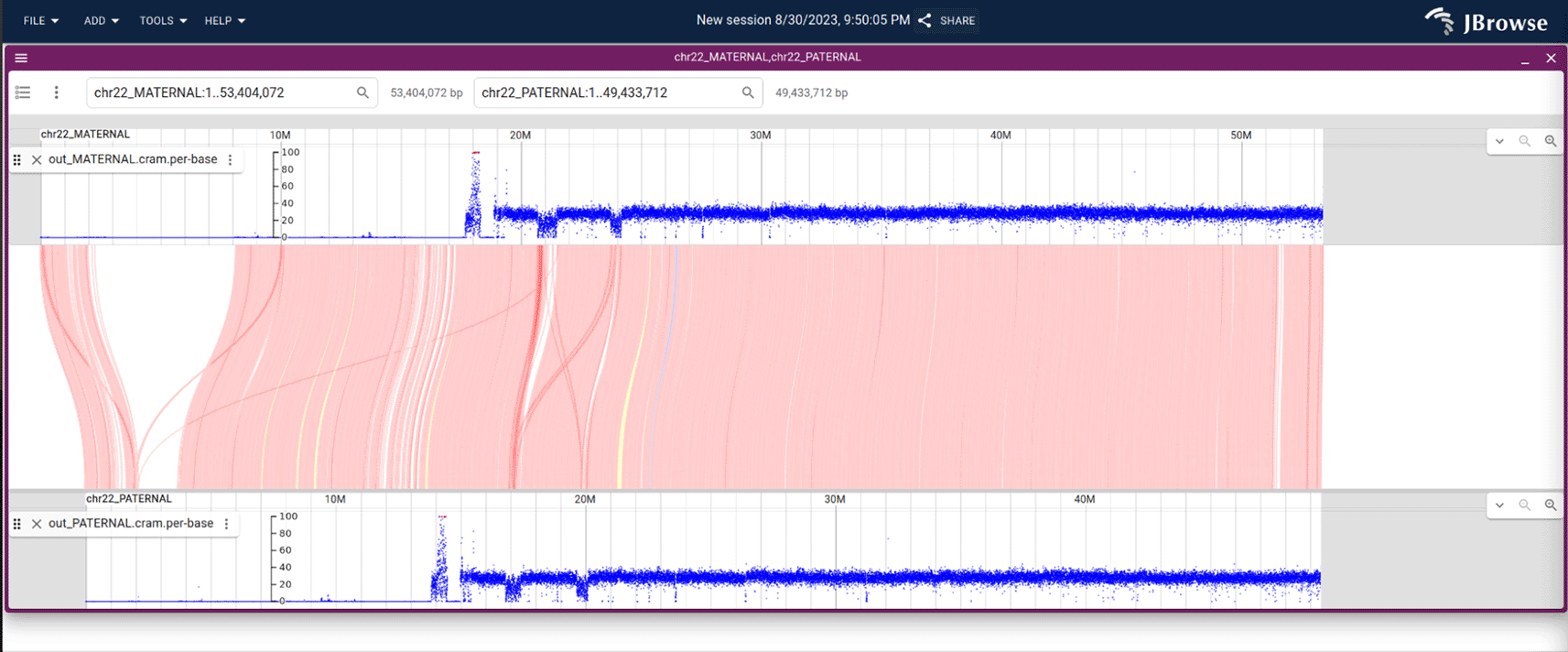
Notably, the maternal chromosome is 4 Mbp longer than the paternal chromosome.
While various tools exist for comparing transcripts, they often overlook the significance of the transcript sequence order. To address this gap, we have devised a prototype algorithm capable of comparing transcripts by considering both their coordinates and sequence compositions. Our ongoing objective is to optimize the algorithm for scalability, with a specific focus on employing memory-efficient techniques suitable for extensive projects, such as the 1000 Genome Project. This optimization will involve refining cluster algorithms and adopting an alignment-free methodology to facilitate transcript comparisons across diverse samples. Presently, our algorithm generates a basic table indicating shared and unique transcripts. However, our ultimate future goal is to exploit this table to provide more comprehensive insights, including information on the spatial relationships between transcripts within each sample. This advancement aims to enhance the depth of analysis and contribute to a more nuanced understanding of transcriptomic data.
SpikeVar and TykeVar successfully enabled creation of simulated genomic data containing known mosaic variants with VAFs between 5% and 20%. To our knowledge, these are the first workflows that simulate mosaic variants for the benchmarking and quality control of mosaic variant callers. The strengths of our workflows include a rapid and reproducible creation of simulated genomic truth datasets with accompanying index VCF files with mosaic variant location and VAF. In addition, both files are widely compatible with mosaic variant callers and provide a variety of different types of variants including SNVs, SVs, and indels and both workflows require only basic packages and are therefore easily installed and implemented.
Benchmarking mosaic variant callers is essential in order to generate reliable data for evaluation of disease associations of mosaic variants. Therefore, we plan to convert both TykeVar and SpikeVar into a one-step tool for the generation of simulated data. We also want to enable the user to have the option to define a global VAF as well as variant specific VAF. In a final step, we will compare our simulated data with data of physically mixed and sequenced samples.
Pangenomes have been used in the past to elucidate the core genomes of pathogens, to improve detection of horizontal gene transfer events, and to study their evolutionary trajectories in different environments. These applications greatly expand our understanding of bacterial and host-pathogen dynamics with practical applications to both medicine and agriculture.
Metagenomics and sequencing of clinical isolates are gaining traction for identification of antimicrobial resistance profiles and diagnosis. Pan-genomics can greatly benefit these clinical applications. Creating pangenomes provides additional insight into pathogen evolution and transmission. For example, including local isolates in pangenomes could inform outbreak investigation efforts and lead to improved infection prevention within hospital systems. Mapping reads directly to pangenomes is a recent advance that may improve detection of polymorphisms related to antimicrobial resistance or virulence. Examining practical considerations and comparison with standard practices demonstrates the promise of alignment to pangenomes and drawbacks.
SalsaValentina enabled identification of putative de novo SVs, two of which were investigated in further detail. One was determined to occur in the T-cell receptor locus, and thus is a likely somatic event that may not be interesting for the use cases of de novo disease associated SVs or variant calling refinement. However, we were able to verify the deletion in HG002 using a local assembly, demonstrating the capability of the pipeline. In future, results could be restricted to certain regions of interest in the genome, excluding known recombination regions. Furthermore, visualization of the candidate de novo SVs could aid in screening candidates in problematic regions or regions of interest. In addition, we observed and confirmed a previously reported de novo SV in HG002, at chr17:51417826–51417932. This variant was identified as part of a comprehensive benchmarking effort for HG002 and thus demonstrates the ability of SalsaValentina to identify genuine events.36 One potential limitation of our local assembly to verify the SV calls is that we used only reads that mapped near the putative SV. In the future, we recommend including unmapped reads in the assembly to ensure reads that failed to map may be incorporated into the contigs.
We set out to fill a critical niche within plant genomics by building a pipeline for analyzing genomes that have not been extensively resequenced, particularly non-model plant systems with limited genomic resources. Our method, PhytoKmerCNV, demonstrates great promise as a reference-free approach for genotyping copy number variation using whole-genome sequencing reads. Moreover, its ploidy-agnostic nature would make it adaptable to genomes with varying levels of ploidy. Such k-mer-based approaches, known for their sensitivity, offer the intriguing possibility of low-pass sequencing, potentially opening up new avenues for exploring CNV dynamics albeit being rather risky. The proposed tool is dependent on the ability to identify reads which match a specific protein domain. Therefore, one of the major risks of this approach is the potential to miss reads originating from sequences of interest but missing the captured protein domain.
Moving forward, several promising improvements can be made. Currently, the existing pipeline generates a k-mer based estimate from the ratio of the sum of k-mers in the NBS-LRR genes captured reads over the sum of k-mers in the total sample. One challenge this approach must overcome is the incredibly high sum of k-mers which return a count of 1. In future hackathons, the sum could be substituted with other summary statistics such as mean, median, and/or statistical methods which account for the inflated counts. Additionally, several other types of gene families could be studied (e.g., transcription factors with specific DNA binding domains, cytochrome P450s, kinase families, heat shock proteins, pathogenesis-related proteins, MADS-box genes, ABC transporters, RNA-binding proteins, and/or late embryogenesis abundant proteins). Each of these have their own pros and cons and must be considered with respect to the type of biological data.
Upon further modification and refinement, we expect the pipeline to generate accurate CNV estimations and help researchers observe the range of variability of CNV across plant genomes. Some genomes may have higher copy numbers of specific NBS-LRR genes, while others may have fewer copies, reflecting the natural genetic diversity within the tomato species. Additionally, an optimized method would allow for further validation of the previously annotated NBS-LRR genes in genome assemblies, and potentially highlight novel gene copies or variants which were not initially annotated. Extending the application of PhytoKmerCNV to a broader range of plant species, especially those with unique genomic characteristics, will enhance its utility in diverse research contexts. Incorporating phenotypic data alongside the CNV analysis can uncover genotype-phenotype relationships, shedding light on the functional significance of CNVs, particularly in the context of disease resistance and other traits.
In the future, we believe that it would be valuable to develop a user-friendly interface and detailed documentation to make the tool more accessible to researchers with varying levels of computational expertise. And finally, we encourage collaboration and feedback from the research community on this particular approach with hopes of fostering improvements and adaptations of PhytoKmerCNV to meet evolving research needs.
SV-Genie provides a generalized framework for evaluating the performance of SV-calling tools. Our analysis shows that SV-calling tools still have a long way to go, since even the best performing SV caller (dysgu46) has a modest recall rate and a high false positive rate, confirming previous reports.46,79 Another issue is that some SV callers (cue80 and SVABA48) have far worse performance with default settings, compared with what was stated in the original publications. In addition, SVABA48 assigns BND as the SVTYPE for all SV calls, but does not provide a script for converting the SV caller output to the standardized VCF format with the standard ‘SVTYPE (e.g., DEL, DUP, INS), while Parliament279 has different chromosome naming conventions. Looking forward, it is strongly recommended that all SV callers should have a minimum set of standardized fields such as SVTYPE, END, SVLEN to make it more straightforward for the end user to use and for SV caller performance evaluation.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)