Keywords
Innate lymphoid cells, Multiple sclerosis, Single-cell RNA sequencing, Transposable elements
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Single-Cell RNA-Sequencing collection.
Innate lymphoid cells, Multiple sclerosis, Single-cell RNA sequencing, Transposable elements
Multiple sclerosis (MS) is a chronic and often immune-mediated demyelinating disease of the central nervous system (CNS) that causes nontraumatic neurological disability in young adults, with a higher incidence in women than men.1,2 Despite extensive research, the causes of MS are not completely understood, but genetic predisposition and environmental factors, such as vitamin D deficiency, obesity, smoking, and viral infection, are thought to influence MS susceptibility and disease progression.3,4 Several viruses have been associated with MS, including herpesviruses, such as Epstein-Barr virus (EBV) and human herpesvirus 6 (HHV-6), and human endogenous retroviruses (HERVs), which are a group of transposable elements (TEs). HERV-W is a type of retrovirus-like particle that has been associated with MS for over 30 years.5–7
TEs are mobile DNAs of viral origin that replicate throughout the genome and compose nearly 50% of the mammalian genome. They act as building blocks for cis-regulatory elements and are categorized into two major classes: retrotransposons, which require RNA intermediates to replicate and re-insert into the genome, and DNA transposons, which directly move through the genome by a cut-and-paste mechanism with the aid of a protein called transposase.8–10 Retrotransposons are further classified into long terminal repeat (LTR) retrotransposons, such as HERVs, and non-LTR retrotransposons, such as long interspersed retrotransposable elements (LINEs) and short interspersed retrotransposable elements (SINEs).11
Recent studies have revealed the role of TEs in fueling genetic innovation in the mammalian immune system as gene regulatory elements. For example, a subset of ERVs has been shown to be involved in regulating vital immune functions, such as the activation of AIM2 inflammasome.12 Additionally, TEs occur more frequently within enhancers that are specific to immune cells. Dysregulation of TEs and subsequent inappropriate immune gene activation may result in various disorders, including autoimmune diseases.13,14
Recent advancements in next-generation sequencing have enabled researchers to uncover the transcriptional state of individual cell gene expression in the subject of interest. Single-cell RNA sequencing has provided novel insight into transcriptional heterogeneity, disease immunology, and diagnosis of various diseases, including neurological disorders.15
CSF surrounds the CNS and contains immune cells, such as central memory CD4+ T cells. CSF is easily accessible and plays a vital role in immunosurveillance, making it a valuable source for studying MS immunology and identifying immune cells involved in MS pathology. However, to the best of our knowledge, no single-cell RNA sequencing studies have identified the possible role of TE dysregulation in cell function and its potential role in MS immune dysregulation.16–18
In this study, we reanalyzed single-cell RNA sequencing data from CSF samples obtained from six MS patients and six patients with idiopathic intracranial hypertension (IIH) as controls to assess the correlation between TE dysregulation and MS pathogenicity.
We retrieved the sc-RNA sequencing data of CSF samples from six MS patients and six patients with IIP as controls from the Gene Expression Omnibus (GEO) database, which can be accessed at https://www.ncbi.nlm.nih.gov/geo/. The accession number for the dataset is GSE138266.
We obtained the aligned sc-RNA sequence reads of the samples directly from the European Nucleotide Archive (ENA) database. To accurately count TEs, we used a recently developed aligner called scTE,19 which is compatible with STARsolo20 output files. This was necessary because conventional aligners typically ignore multi-mapping reads, which can be critical for counting TEs. The ENA database is a publicly accessible repository of DNA and RNA sequence data, while scTE is a specialized aligner designed specifically for counting TEs in single-cell RNA sequencing data. STARsolo is a popular tool for processing sc-RNA sequencing data, which generates output files that can be used with scTE.
We built genome indices using the scTE-build function, which utilizes the UCSC genome browser Repeatmasker track21 and the GENCODE database in default mode (exclusive). We then used the scTE function to realign the BAM files and generate count matrices for further investigation. The count matrices generated by scTE provide a quantitative measure of the expression levels of TEs in single-cell RNA sequencing data. This approach allows for accurate counting of TEs in multi-mapping reads, which is critical for understanding their potential role in disease pathology.
We used the Seurat R package,22–24 which is a powerful tool for analyzing single-cell RNA sequencing data and visualizing results. To preprocess the data, we first filtered out low-quality cells and cell doublets based on the following criteria: 1) cells with greater read-counts RNA than 98% of cells in the sample, 2) cells with low numbers of genes (less than 2% of cells in each sample), or 3) cells with more than 40% mitochondrial counts. The data were then normalized using the ‘LogNormalize’ method, and the 2000 most variable genes were identified using Seurat’s ‘FindVariableFeatures’ function. To correct for batch effects, the samples were integrated using the ‘IntegrateData’ function in Seurat. This allowed us to combine the scRNA-seq data from the different samples and reduce technical variability, making it easier to clustering the MS and control samples.
To ensure that TEs did not affect our clustering analysis, we temporarily removed them from the count matrix and performed data scaling and PCA using Seurat’s ‘RunPCA’ function with default parameters. Next, we generated a KNN graph based on the PCA-reduced data and performed unsupervised clustering using the ‘FindNeighbors’ and ‘FindClusters’ functions in Seurat. After clustering the cells based on their gene expression profiles, we added the TEs back into the count matrix using their cell barcodes. This allowed us to identify cell subpopulations based on their gene expression patterns and investigate the expression of TEs within these subpopulations.
To identify differentially expressed genes, we performed differential expression (DE) analysis on the ‘RNA’ assays of the count matrix using the ‘MAST’ method25 in Seurat’s ‘FindAllMarkers’ function. We also used canonical cell markers to identify the cell type of each cluster. DE analysis allowed us to identify genes that were significantly differentially expressed between clusters.
To investigate the potential association of upregulated TEs in the cluster of interest with transcription factors (TFs), we queried each TE with a log fold change (log FC) greater than 0.9 and an adjusted p-value less than 0.05 in the RTFAdb database.26
We then obtained a list of all TFs with a possible association with upregulated TEs and performed gene set enrichment analysis using the Enrichr web server.27,28 Enrichr is a powerful tool that allows for the analysis of gene sets and the identification of biological pathways and processes that are overrepresented in a given gene list. By performing this analysis, we were able to gain additional biological insights into the potential role of TEs in the biological functions of the clustrer of intrest.
To determine the discrete immune cell populations and the potential role of TEs in MS pathogenicity, we reanalyzed single-cell RNA sequencing (scRNA-seq) data from cerebrospinal fluid (CSF) samples obtained from MS patients and patients with idiopathic intracranial hypertension (IIH) as controls. The scRNA-seq dataset included a total of 23,423 cells from MS patients and 19,725 cells from controls.17 ( Table 1)
We employed the k-nearest neighbor (KNN) graph constructed on the principal component analysis (PCA)-reduced data to cluster single cells and then labeled each cluster based on canonical cell markers (Supplementary Table 1). The average and percentage expression of each canonical cell marker in MS and IIH patients are depicted in Supplementary Figure 1 and Supplementary Figure 2. We also utilized uniform manifold approximation and projection (UMAP) plots to visualize the clusters in the MS and IIH populations (Figure 1). Based on the canonical markers, we identified major clusters including T cells, innate lymphoid cells (ILCs), natural killer (NK) cells, B cells, plasma cells, CD14+ monocytes, dendritic cells, and plasmacytoid dendritic cells.22,29
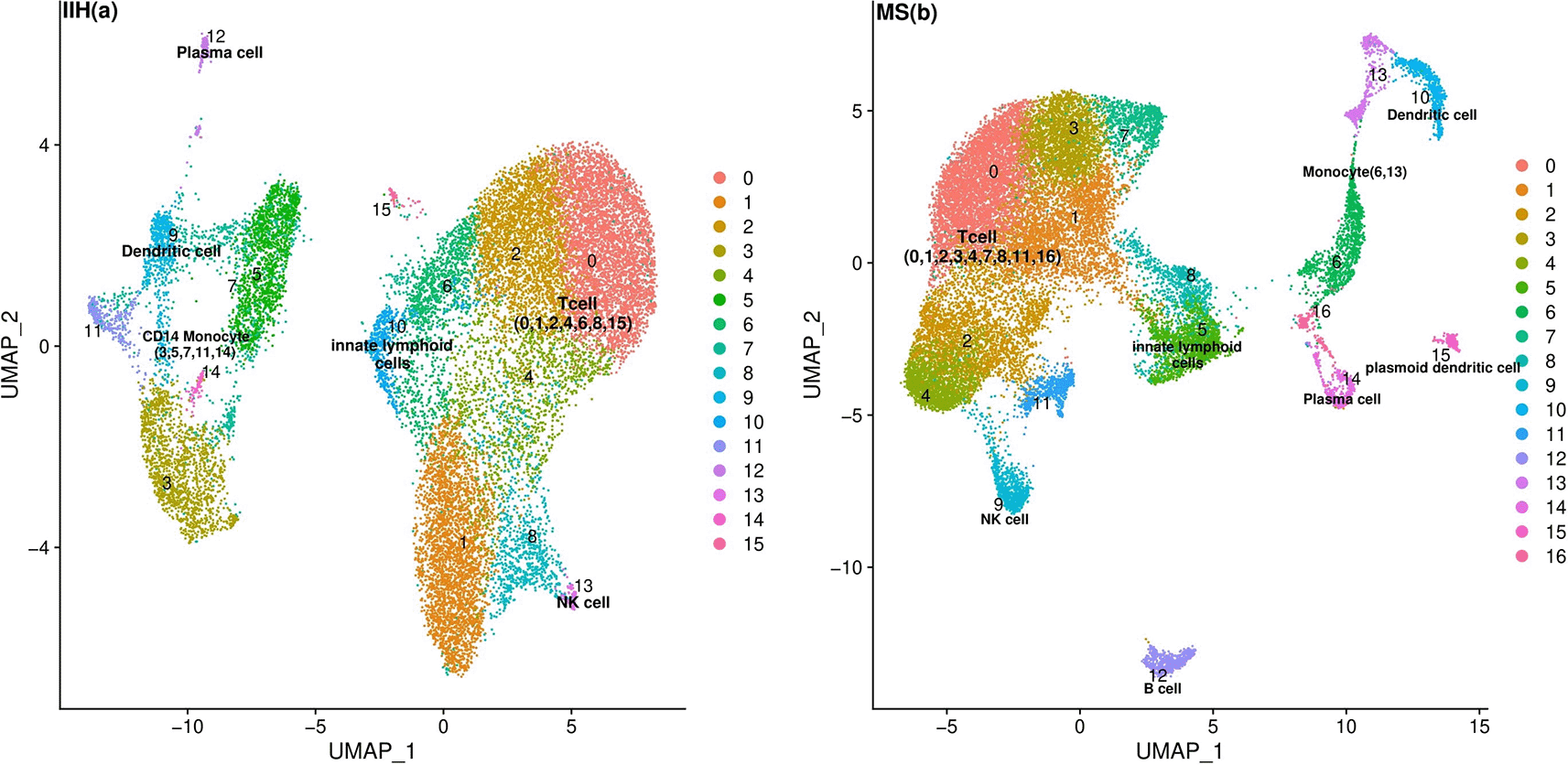
The plot also highlights the clusters associated with the identified major cell types. Each cluster is represented by a specific color and number.
Among all these clusters, ILCs revealed a distinct pattern for TE upregulation (Supplementary Table 2 and Supplementary Table 3). This cluster, differentially expressed a high number of TEs (logFC > 0.5 and adj. p-value < 0.05) (Figure 2-a and Figure 2-b). Also, this cluster had a higher frequency in MS (Ctrl n = 485 vs MS n = 1548 ILC cells) (Supplementary Table 4 and Supplementary Table 5).
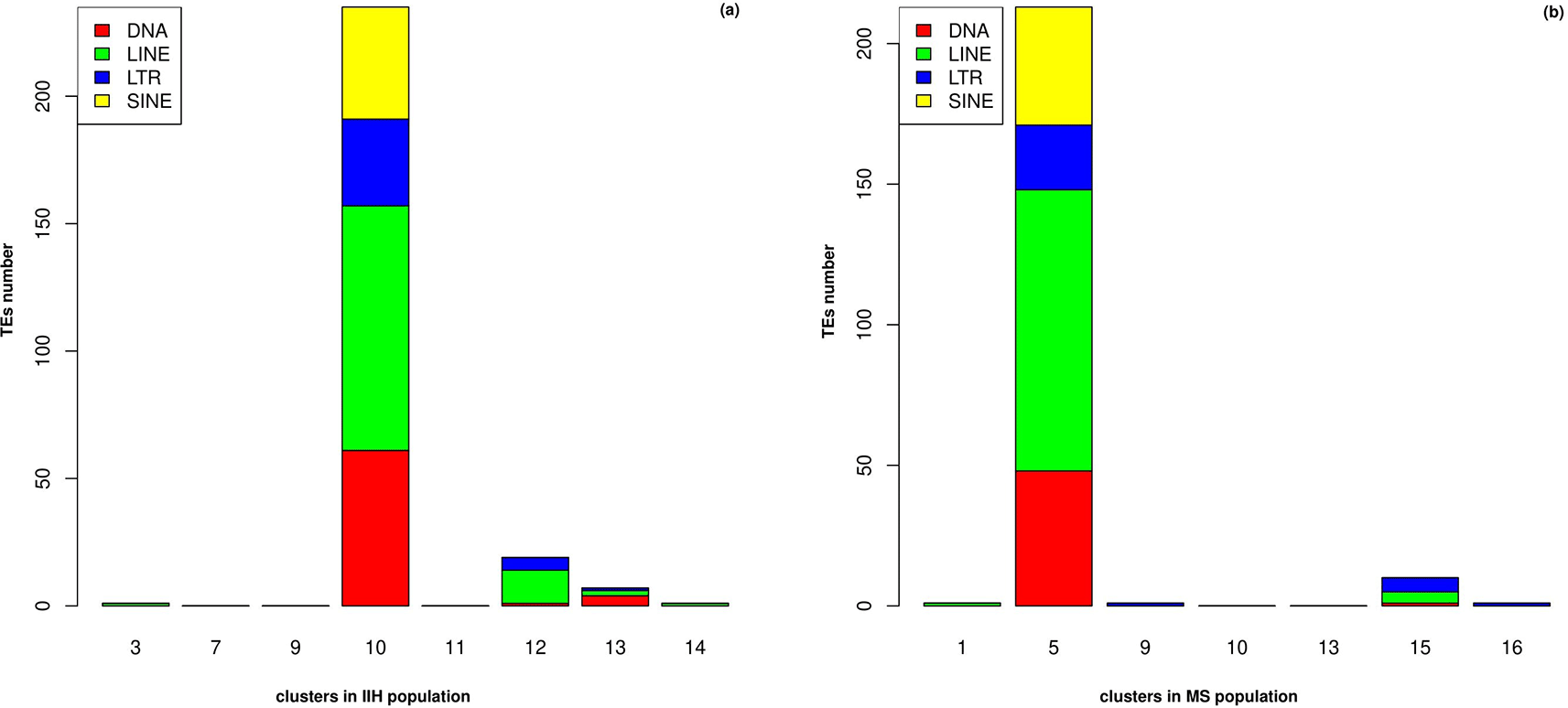
The length of each bar represents the number of upregulated TEs from each subfamily.
Various defense mechanisms, such as DNA methylation and histone modification, can suppress TE activity.30 Given that TEs are upregulated in the ILC cluster, it is reasonable to assume that these mechanisms may be reduced in ILCs. To identify the potential role of TEs in influencing the transcriptional activity of nearby genes by providing binding sites for transcription factors (TFs), we analyzed the association of upregulated TEs (logFC > 0.9 and adj. p-value < 0.05) with TFs using the retrotransposon-transcription factor associations database (RTFAdb).26 We identified 54 TFs that could potentially bind to TE-enriched locations in the genome and affect the ILCs’ transcriptional regulatory network (Supplementary Table 6). The enrichment analysis of the associated TFs with TEs is shown in Figure 3. Cell-type enrichment analysis confirmed the association of the identified TFs with immune cells.22 Moreover, the enrichment analysis indicated that the selected TFs were most significantly associated with immune pathways, including the TNF and IL-17 signaling pathways.31,32 Furthermore, gene ontology (GO) molecular function enrichment analysis revealed the association of the selected TFs with cis-regulatory region sequence-specific DNA binding, which supports the potential role of TEs in providing cis-regulatory sequences in the genome.33,34
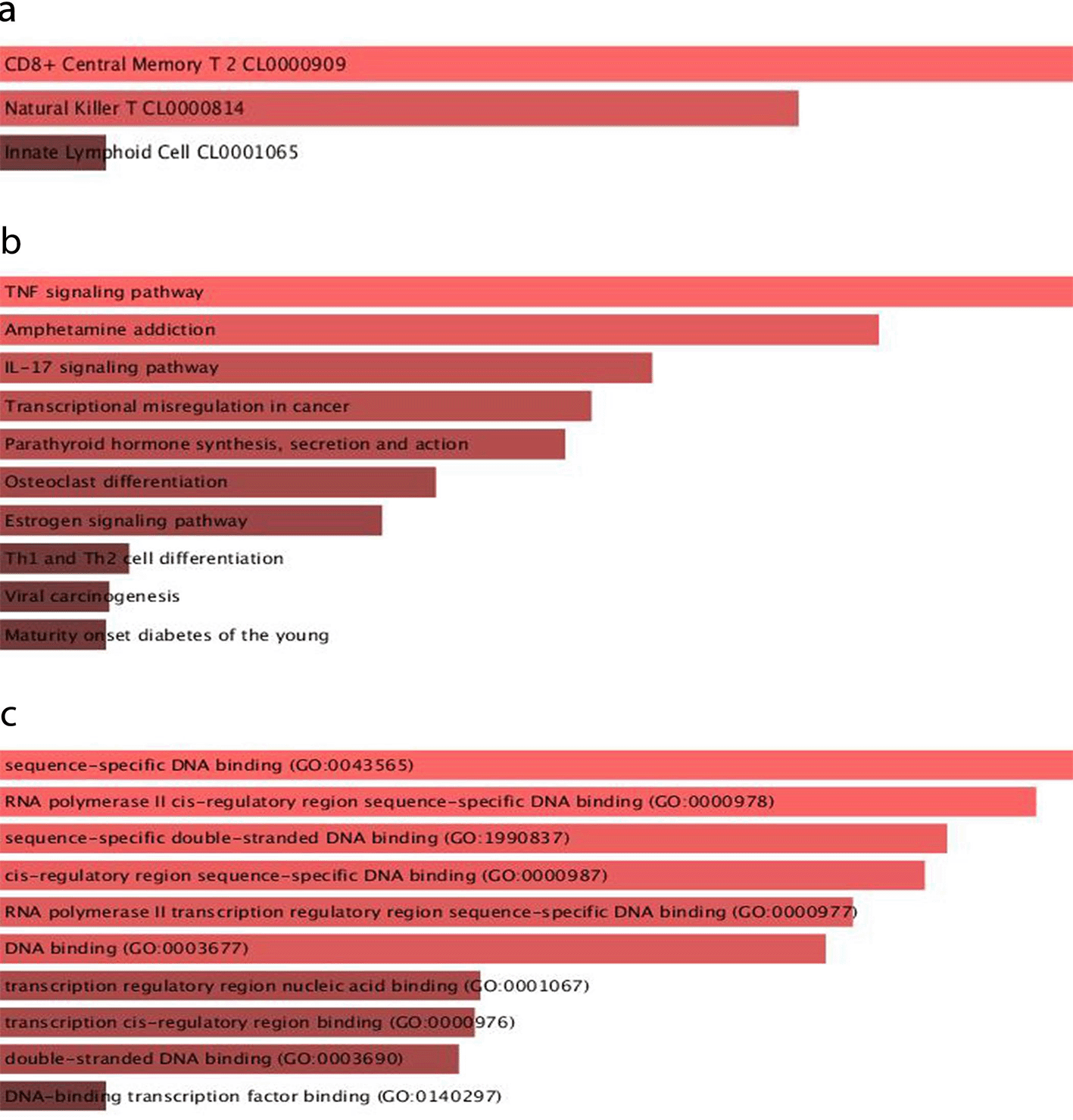
(a) Azimuth cell types_2021 (b) KEGG 2019 (c) GO molecular function_2021.
In this study, we aimed to uncover the potential role of TEs in MS pathogenicity. We identified a cluster of ILCs with a higher population in MS patients than in the control group. ILCs are non-T cell, non-B cell, and NK cell-like lymphoid cells that belong to the innate immune system.35 ILCs are categorized into different subsets, including NK cells, lymphoid tissue inducer cells (LTi), and helper ILC1-3.35 Recently, ILCs have been considered neglected players in MS pathogenicity.36 We identified that TEs are highly upregulated in ILCs. This TE upregulation in ILCs may be in accordance with the ability of these cells to adopt their phenotype and functional capacities in response to microenvironment changes, a phenomenon called plasticity.36,37 It has been proposed that ILC plasticity requires defined chromatin regions, such as cis-acting enhancers and silencers of gene expression, to be accessible to TFs.38 Since TEs are repetitive and mobile in nature, one can speculate that TEs are involved in the genomic plasticity of ILCs by inserting themselves in coding or regulatory regions of immune-related genes, subsequently impacting gene expression and inducing plasticity.39
It has been revealed that the proportion of LTis was significantly increased in the CSF of patients with early MS, suggesting either recruitment of blood-derived ILCs or a local expansion of ILCs in the CSF.40 Additionally, it has been shown that a subset of blood-derived inflammatory ILC3s could develop MS-like disease in mouse models by infiltrating the CNS and presenting the antigen to myelin-specific T cells.41 Many studies have revealed that TEs are overexpressed in inflammatory and immune-mediated diseases.11,42,43 For instance, TE upregulation is observed in many cancers due to DNA methylation loss in cancer cells. This can lead to the presentation of TE-derived peptides on MHC-1 molecules that are recognized by T cells, resulting in immune cell infiltration and inflammation.44
ILCs could directly augment T-cell adaptive immunity by upregulating the expression of MHC class II, especially in inflammation. One may speculate that this process could occur in immune-mediated diseases.45 Therefore, we propose that presenting TE-derived antigens by ILCs with overexpressed TEs could reactivate T cell-mediated immunity and lead to subsequent MS progression. Consistent with this assumption, antiretroviral agents that can suppress TE expression have demonstrated potential therapeutic benefits in MS treatment.46–52
The detrimental role of TE activity in neurodegenerative diseases has also been revealed by several studies.53–56 The most common explanation for TEs’ role in neurodegenerative diseases is an inflammatory response to double-stranded RNA (dsRNAs) and/or cDNAs, or a direct cytotoxic response to specific TE-derived proteins which are mainly directed by innate immune responses.57 In this regard, it has been revealed that peripheral blood mononuclear cells (PBMCs) obtained from MS patients induce elevated Interferon-gamma (IFNγ), Interleukin 6 (IL-6), and tumor necrosis factor-alpha (TNF-α) expression after the stimulation of these cells with multiple sclerosis-associated retrovirus element (MSRV) Env protein. It appears that HERV-W Env proteins activate innate immune responses by activating toll-like receptor 4 (TLR4) and CD14, resulting in substantial proinflammatory cytokine production.11 The common hypothesized model of retroelement-mediated neuroinflammation proposes that the resultant proinflammatory cytokines produced by the detection of retroviral-derived particles by innate immune response receptors could exert neuropathophysiological effects.11 However, in this study, we propose a new possible mechanism that is mediated by functional interactions between ILCs and adaptive immunity in MS pathogenicity.
In this study, we reanalyzed the single-cell RNA sequencing data from CSF samples obtained from six MS patients and six controls to assess the correlation of TEs with MS pathogenicity. We found that ILCs expanded in MS revealed a distinct pattern of TE upregulation. The upregulation of TEs in these cells is consistent with the adaptive phenotype and functional capacities of these cells in response to stimuli.37,58 It is possible that the genomic plasticity of ILCs could be obtained by the insertion of TEs in coding or regulatory regions of immune-related genes, leading to functional impacts on gene regulation and subsequent plasticity.39 The enrichment analysis of the associated TFs with highly upregulated TEs in ILCs revealed the relevance of the TFs with immune pathways and cis-regulatory regions in DNA, which supports this hypothesis.
Additionally, we proposed a new hypothesis regarding the possible role of TEs in MS pathogenesis. The main model for TEs’ role in neurodegenerative diseases is based on the sensing of TE-derived particles by pattern recognition receptors in innate immune response and the subsequent inflammation and neurodegeneration.11 However, we hypothesize that presenting TE-derived antigens by ILCs with overexpressed TEs could reactivate T cell-mediated immunity and subsequent MS progression. Based on this assumption, the administration of antiretroviral agents that can suppress TE expression could be used in MS treatment.51,53 Further studies are needed to investigate the probable role of TEs in neurodegenerative diseases’ pathogenicity and their relevance to immune system plasticity. Further studies are needed to investigate the role of TEs in neurodegenerative diseases’ pathogenicity and their relevance to immune system plasticity. Additionally, it seems that repurposing nucleoside reverse transcriptase inhibitors (NRTIs) or developing new high-efficacy NRTIs that specifically target retrotransposon reverse transcriptases would be a feasible approach in MS treatment.59
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)