Keywords
Direct repeats, DNA repair mechanisms, R package, repeat annotation
This article is included in the Genomics and Genetics gateway.
This article is included in the RPackage gateway.
Direct repeats, DNA repair mechanisms, R package, repeat annotation
The discovery of repetitive DNA sequences through reassociation experiments marked a foundational moment in genome biology.1 However, the annotation of repetitive elements remains technically challenging, and despite their widespread presence, our understanding of their functional impacts continues to lag behind that of more well-characterized genomic features. In part, repeat annotation is hampered by the sheer diversity of repetitive sequences. Repeats can occur as long tandem arrays, others as dispersed copies, and they span a wide range of sizes, sequence identities, and evolutionary origins.2
These repeat architectures have variable impacts on genomes and are now recognized for their roles in genome stability/instability, evolution, and disease.3–5 Repeats can influence mutation and recombination rates,6,7 and their expansion/contraction underlie human disorders like Huntington’s disease and fragile-X syndrome.8 Additionally, microsatellites have been shown to evolve rapidly and vary considerably in abundance even among closely related taxa, underscoring their dynamic evolution9; they also contribute to epigenetic regulation and can influence gene expression by activating promoters or serving as transcription factor binding sites.10,11
Proximal direct repeat pairs are of particular interest. These are two identical sequences in the same orientation, separated by a spacer region. In both bacteria and mammals, the distribution and abundance of long direct repeats appear to be shaped to minimize genome instability, with constrained chromosomal positioning in bacteria12 and a potential role for natural selection in reducing repeat-mediated mutagenesis in the mitochondrial DNA of longer-lived mammals.13 This mutagenic potential is also evident at the molecular level, where direct repeat pairs facilitate the DNA repair pathway single-strand annealing (SSA) (Figure 1). After a double-strand break occurs, the 5’ ends of each strand are resected, and complementary repeats are used to anneal the strands. The repair mechanism is considered mutagenic because the intervening DNA between repeats and the downstream repeat is lost during the repair process.14
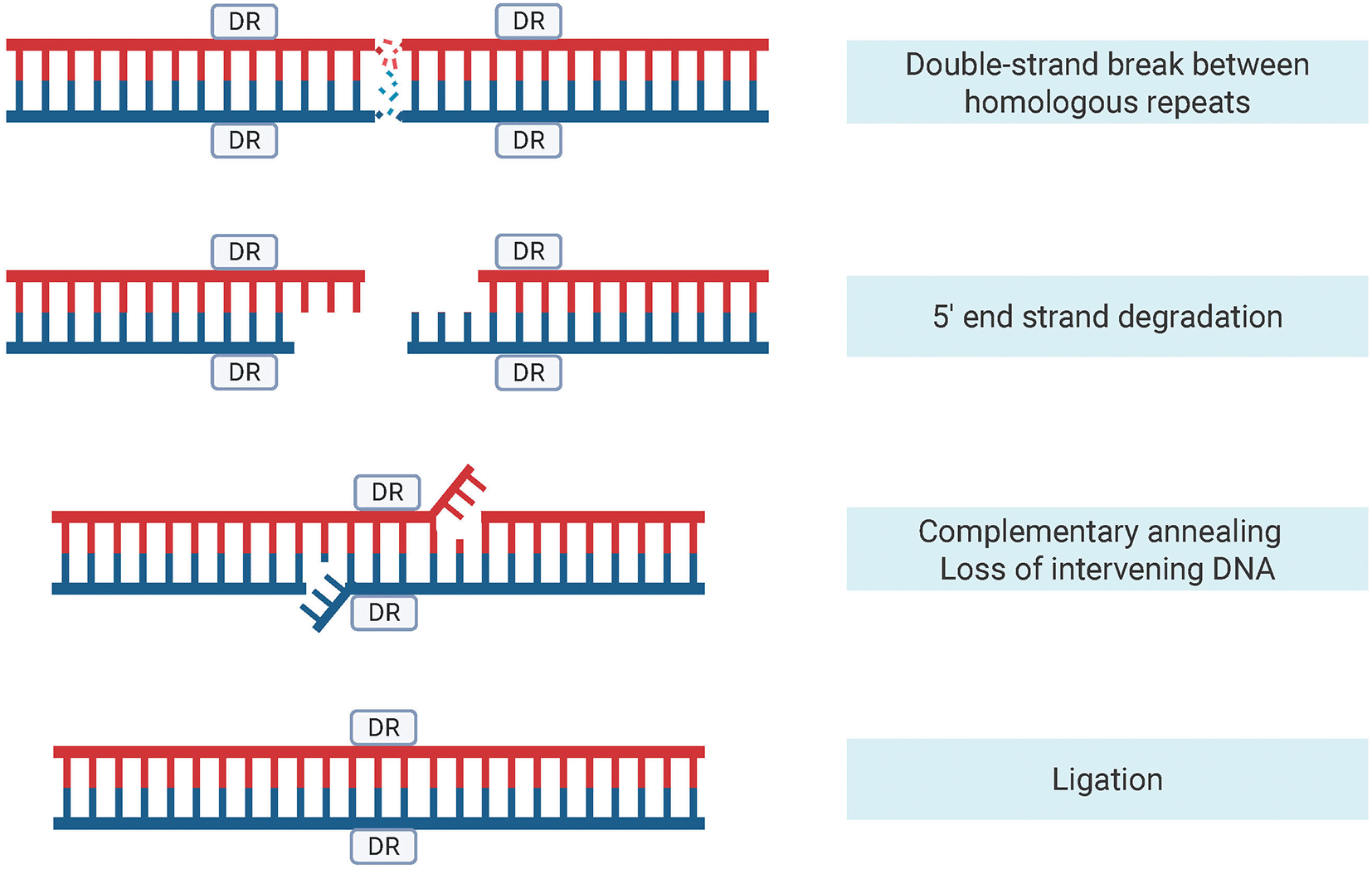
This pathway is initiated by a double-strand break between homologous DNA repeats followed by the resection of the 5' end strands, creating 3' overhangs. These overhangs find and anneal to complementary sequences, resulting in the loss of intervening DNA and the downstream repeat. The final step is ligation, where the DNA strands are rejoined, completing the repair process. Created in BioRender. Copeland, M. (2025) https://BioRender.com/d9g3lm9.
Tools such as RepeatMasker,15 HelitronScanner,16 ab initio programmes like PILER,17 and combinatorial pipelines such as RepeatModeler18 are widely used to perform repeat annotation or classification. More recently, Repeater19 introduced fast, alignment-free profiling of diverse repeat classes and produces informative whole-chromosome visualisations. While these tools are well suited for identifying broad repeat families or consensus-based repeat structures, they are not able to pinpoint exact, spatially localized repeat pairs and provide detailed annotation information for the repeats.
Here we present DirectRepeateR, an open-source R package that fills this gap. DirectRepeateR performs de novo identification of exact direct repeat pairs within a user-specified distance and immediately returns analysis-ready CSV files or optional GFF files. DirectRepeateR is implemented entirely in R, and all user interaction remains within the R environment, making it easily accessible to researchers with minimal coding experience. DirectRepeateR offers a complementary approach by focusing specifically on the identification of exact direct repeat pairs at user-defined spatial and length resolutions. This fine-scale, length-consistent detection is particularly useful for studying localized structural features, such as repeat-mediated recombination or deletion events.
To illustrate its use, we map direct repeat pairs in the Aedes aegypti genome and, using a Monte Carlo simulation, test whether the distribution of direct repeat pairs in Aedes aegypti is consistent with selection against their presence flanking exons.
The DirectRepeateR package
DirectRepeateR is an R package designed to identify, annotate, and visualize nearby direct repeat sequences within genome assemblies. It offers a flexible solution for detecting direct repeats using a de novo approach, making it useful for model and non-model organisms. This package features three functions: GetRepeats, ConvertToGFF, and PlotRepeats. We made a vignette that provides guidance on how to use each function (S1).
GetRepeats function
The function GetRepeats is designed to identify direct repeat sequences from genome assemblies, leveraging R for a simple user interface and C++ for efficient processing. While this package incorporates a C++ backend for performance, all user interaction takes place through the R interface, keeping the software user-friendly and accessible to researchers with basic R skills. The function takes a genome assembly in FASTA format as the input along with the parameters query_length (length of the substring used to search for repeats), maxdist (the window size in which we search for repeats), and minlength (the minimum length to be considered a repeat). These parameters allow user control over the query sequence length, the maximum distance between repeat copies, and the minimum repeat length, respectively. If these parameters are not provided, default values are used (query_length = 25, maxdist = 20,000, and minlength = 50). These default settings are based on common expectations about the repeat structures targeted by the SSA pathway.14
The function begins by using the C++ backend function through the Rcpp package.20 For each sequence in the FASTA file, this C++ function extracts chromosome lengths and names from the genome and uses these to organize the repeat information. The implemented algorithm is based on sliding across the genome in steps equal to the query length. For each query, the algorithm searches for all exact matches within the range defined by maxdist, representing the maximum allowable distance between repeat copies. This process is repeated until the end of the sequence is reached. When matches are found, the start and end positions of the first element and the match are recorded. As this process is completed for each sequence in the FASTA file, data frames containing the position information are written out as temporary chromosomal CSV files. After the C++ routine completes, GetRepeats processes all the generated chromosomal CSV files inside of R. These temporary files are processed using the minlength argument and are combined to provide the final comprehensive list of detected direct repeats, including their start and end positions along with the positions of their corresponding matches.
ConvertToGFF function
The ConvertToGFF function is designed to convert repeat data into a GFF (General Feature Format) file, a standard format for describing genes and other features in genomes. The user provides the data frame, provided by the GetRepeats function containing the identified direct repeats, and the function works by preallocating vectors to store the GFF entries, including fields for chromosomes, source, feature types (repeat_region for the full length and repeat_unit for individual copies), start and end positions, and attributes. Each repeat in the data frame is processed to generate three GFF entries: one for the full repeat region and two for the individual elements of the repeat.
PlotRepeats function
The PlotRepeats function uses the ggplot2 package21 and generates visualizations of repeat densities across chromosomes using a sliding window approach. This function uses the data generated by the GetRepeats function and allows users to specify window and step sizes (defaulting to 200 Kb for both if not provided). The function processes each chromosome by first calculating the midpoint of each repeat in the file and then sliding windows across the chromosome length. For each window, it counts the number of repeats in the window. It then generates a plot of repeat density along the chromosome.
The DirectRepeateR package requires Rcpp (>= 1.1.0), and therefore requires a version of R >= 4.4.1 (available from www.r-project.org). Users can use the devtools package to install DirectRepeateR from GitHub (https://github.com/coleoguy/DirectRepeateR).
To illustrate the use of our package, we analyzed the A. aegypti genome (GCF_002204515.2) with DirectRepeateR. The A. aegypti genome is approximately 1.3 Gb in size, with 65% being repetitive content.22 We used the DirectRepeateR package to map the location of direct repeats within this genome. For this, we define direct repeats as sequences that have a minimum element size of 50 bp, are oriented in the same direction, and are separated by no more than 20 Kb.
We then used the output from DirectRepeateR to explore whether the distribution of direct repeats was consistent with the hypothesis that selection should limit their proximity to exons. For this project, we considered an exon flanked if it was either spanned or overlapped by direct repeats. Essentially, any orientation where single-strand annealing would be expected to lead to the loss of exonic DNA was considered a flanking arrangement.
We calculated the observed and expected numbers of exons flanked by direct repeats in the A. aegypti genome. To get the observed counts, we used the direct repeat information produced by DirectRepeateR and the annotation file from NCBI (GCF_002204515.2). The GFF file was filtered to retain only unique protein-coding exons, and these exon locations were then compared with repeat locations to identify exons flanked by repeats. To determine expected counts, we employed a Monte Carlo simulation to create a null distribution of the number of flanked exons. We generated direct repeat positions by randomly sampling locations based on the length of each chromosome while maintaining the size of each repeat copy and the distance between copies. When the gap between repeats was under 26 bp, the subsequent repeat was placed by offsetting from the repositioned end of the prior repeat by the same gap, maintaining clustering. After positioning the repeats randomly, we used the same method described above to evaluate the number of flanked exons. This process was repeated 100 times to generate a null distribution. While not included in the DirectRepeateR package, the scripts used for this analysis are provided in a GitHub repository for researchers to use and adapt for their own needs. AI was used to revise software and analysis code to maximize efficiency.
We assessed the runtime of our repeat detection algorithm across a range of species with varying genome sizes. We used the genomes of Caenorhabditis elegans (GCF_000002985.6), Anopheles gambiae (GCF_943734735.2), Vitis vinifera (GCF_030704535.1), Oryzias latipes (GCF_002234675.1), Gallus gallus (GCF_016699485.2), Aedes aegypti (GCF_002204515.2), Mus musculus (GCF_000001635.27), and Homo sapiens (GCF_000001405.40), which span genome sizes from 100.3 Mb to over 3.1 Gb (Figure 2). Runtime scaled linearly with genome size (slope = 0.0677, R2 = 0.96, p = 2.33 × 10−5), indicating a strong relationship with minimal residual variation. This supports the consistent performance of the method across a wide range of genome sizes. Smaller genomes, such as C. elegans (100.3 Mb), completed in under five minutes, while the largest genome, H. sapiens (3.1 Gb), required approximately 3.6 hours.
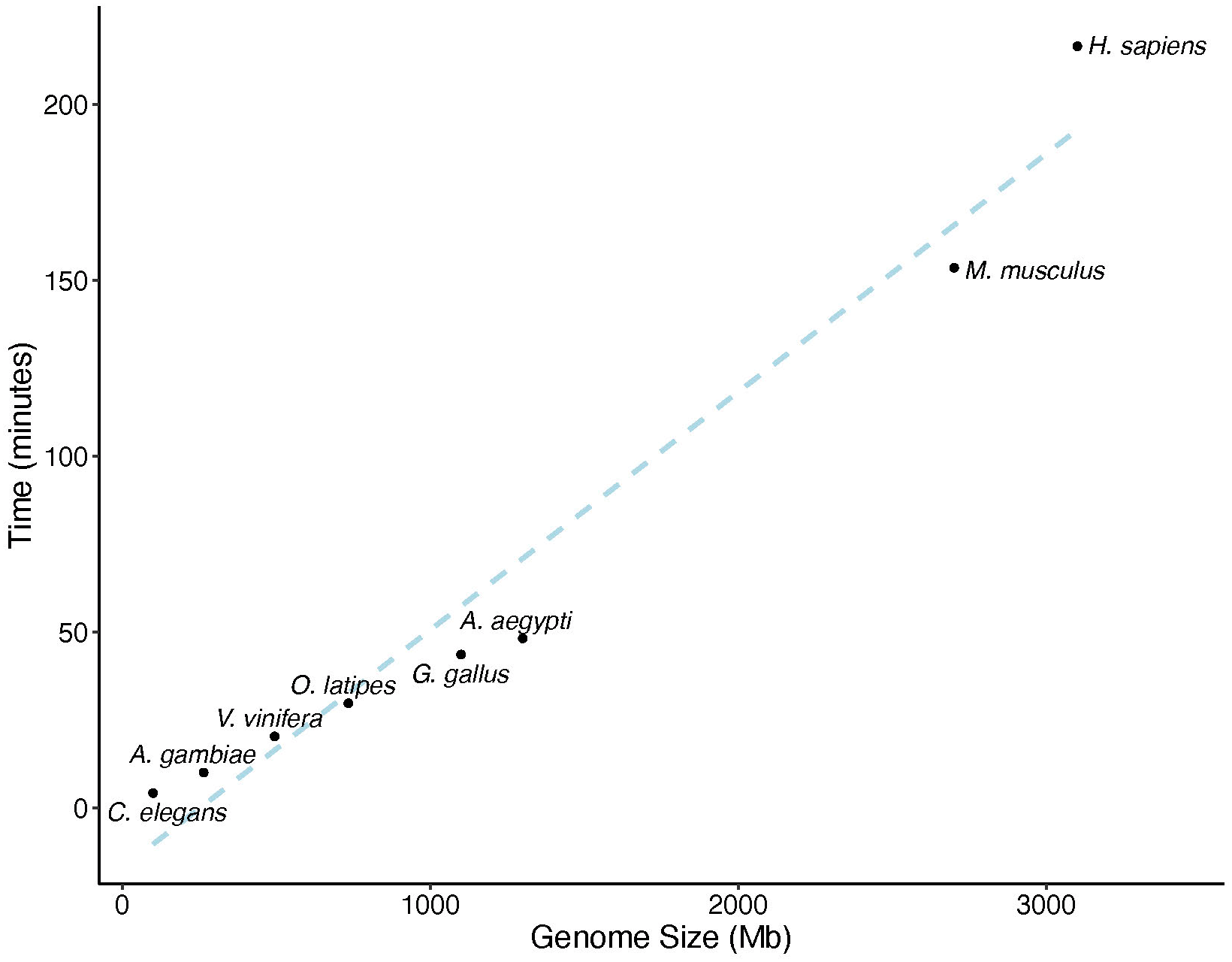
The plot shows the relationship between genome size (in megabases, Mb) and runtime (in minutes) for eight species. The blue trend line indicates linear regression (R2 = 0.96).
In the A. aegypti genome, we observed that 5,782 out of 80,498 exons were flanked by direct repeats. The expected number, based on simulations, was calculated as just under 40,000, highlighting a significant deviation from what our null model predicted (Figure 3). When we analyzed the data by chromosome, chromosome 1 had 1,247 flanked exons, chromosome 2 had 2,427, and chromosome 3 had 2,108 (Figure 3). In all cases, we found that observed values were markedly lower than the expected numbers, with the simulations predicting nearly 14,000 flanked exons for chromosomes 2 and 3, and approximately 10,000 for chromosome 1. These results reveal a consistent pattern across the genome, where the number of exons flanked by direct repeats is far below random expectation.
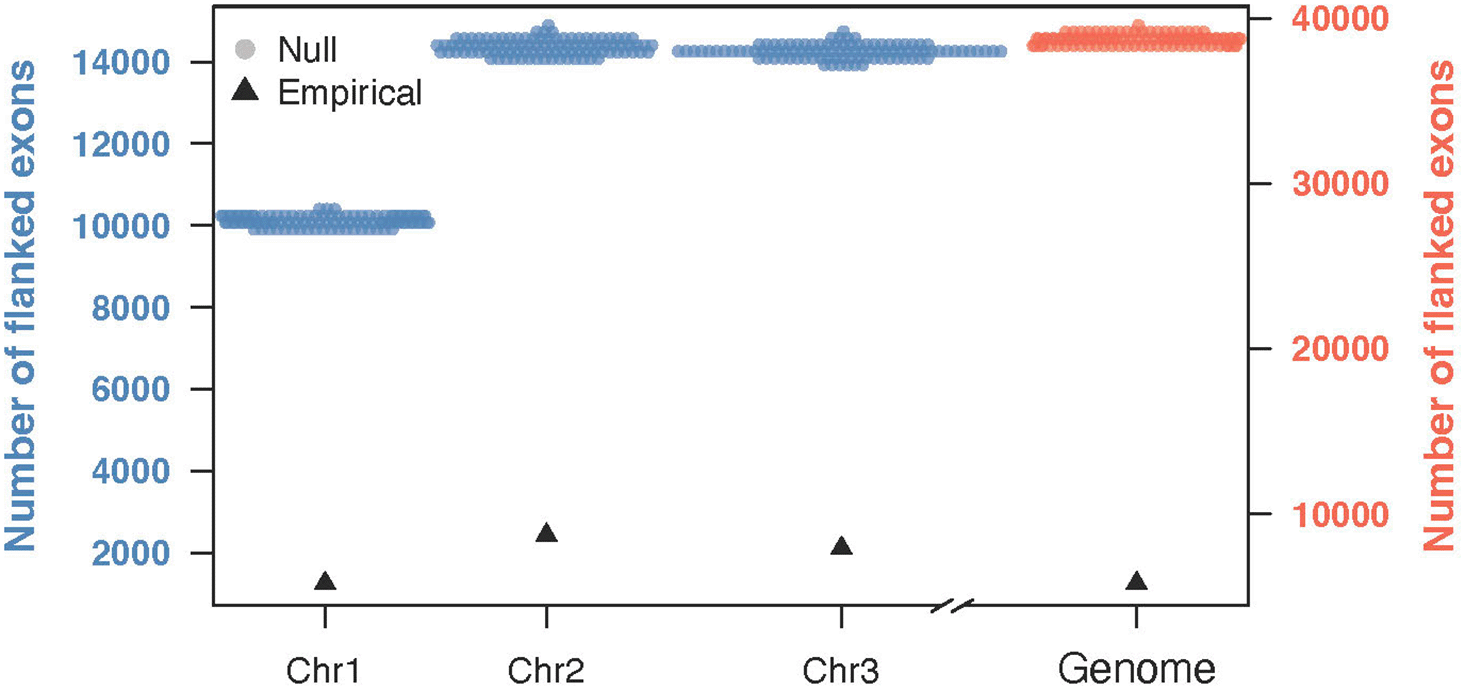
The null distribution (colored points) represents the expected number of flanked exons under a random model, while the empirical values (colored points with black border) show the actual observed counts. Each chromosome and the genome-wide totals are presented separately. The left y-axis (blue) corresponds to chromosome-level comparisons, and the right y-axis (orange) represents genome-wide values.
To investigate the relationship between gene density and repeat content, we analyzed the distribution of genes and direct repeats across 200 Kb windows along three chromosomes of Aedes aegypti. For each chromosome, gene and direct repeat counts were calculated within each sliding window. To visualize the data, we created a scatterplot of repeat counts on a log scale against gene counts. Fitting a linear model to this data showed a significant negative relationship (slope = -10.34, R2 = 0.006, p-value = 1.9e-09), suggesting that regions with higher gene densities tend to have lower numbers of repeats (Figure 4).
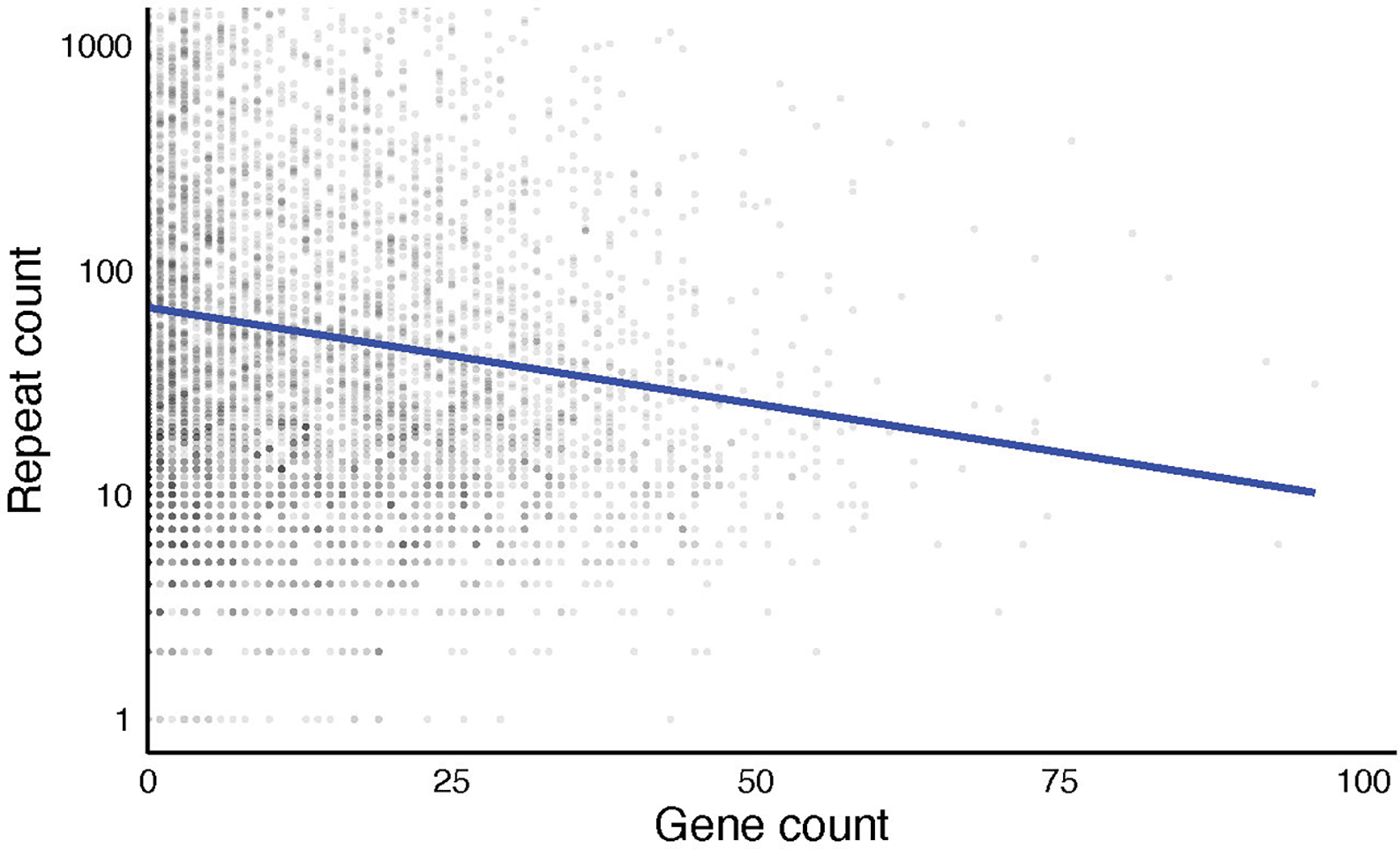
To improve visualization, repeat counts were log-transformed prior to plotting; however, the vertical axis was back-transformed to ensure values remain directly interpretable. Each point represents a window, with the horizontal showing the gene count per window and the vertical axis showing the repeat count. The blue trend line indicates linear regression (R2 = 0.006).
Our analysis shows that exons are flanked by proximal direct repeat pairs less frequently than expected in A. aegypti, and repeat density decreases in gene-rich regions. Since direct repeats can cause deletions via the SSA pathway, their depletion near genes is likely due to purifying selection acting to minimize mutational risk. Across multiple species, purifying selection appears to act on repeats. Studies have suggested that selection may limit TE accumulation near genes,23 restrict TEs to AT-rich, gene-poor regions,24 and constrain microsatellite variation due to its potential to disrupt gene function.25 Together, these findings support the view that purifying selection acts broadly across genome landscapes to restrict repeats from genic regions where their expansion or instability would be most harmful.
The current version of DirectRepeateR is intentionally streamlined for one very specific task—detecting exact, proximal direct repeat pairs. As such, several broader repeat-analysis features are outside its present scope. First, the algorithm currently searches for perfect matches in the same 5′→3′ orientation. The package does not yet identify degenerated, inverted, or mirror repeats. Additionally, DirectRepeateR identifies exact matches by stepping across the genome in increments equal to the specified query length (e.g., 25 base pairs), which can result in missing some portion of the start or end of a repeat if the window does not align perfectly with the repeat’s boundaries. Specifically, this approach may miss anywhere from zero base pairs to the query length minus one at both the beginning and the end of each repeat. This number of base pairs lost from each repeat is uniformly distributed given that repeats can start at any arbitrary position along the genome. This limitation creates a trade-off between runtime and completeness. A smaller query length increases the number of iterations and runtime but improves completeness by aligning the window more accurately with repeat boundaries, reducing missed base pairs. A larger query length decreases runtime by reducing iterations but may miss more base pairs, compromising completeness. These constraints reflect deliberate design choices that prioritise computational efficiency and user-ease for direct repeat studies, but they also highlight clear avenues for future development, most notably adding support for near-identical repeats and inverted orientations.
DirectRepeateR offers a targeted and accessible approach for identifying exact, proximal direct repeat pairs in genome assemblies. Researchers can fine-tune resolution and spatial sensitivity using adjustable parameters, such as maximum repeat distance, to detect repeat structures relevant to genome stability. Fully implemented in R, the package provides a lightweight interface suitable for users with minimal coding experience. DirectRepeateR complements existing repeat-annotation tools by filling a practical gap in the detection of spatially localized, length-consistent repeat pairs.
Code for analysis available from: https://github.com/coleoguy/DRproj26
Archived source at time of publication: 10.5281/zenodo.17073598
This project contains:
• scripts/* (R scripts for repeat annotation and observed flanking exon analysis)
• results/* (Outputs from the R scripts including repeat counts and flanked exon results)
• figures/* (Visualizations generated for the manuscript)
License: Creative Commons Attribution 4.0 International (CC-BY 4.0)
Source code available from: https://github.com/coleoguy/DirectRepeateR27
Archived source at time of publication: 10.5281/zenodo.16275409
License: OSI approved open license software is under MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)