Keywords
subarctic soil, predatory bacteria, Myxococcota, Polyangiaceae, Mucilaginibacter cryoferens, short-read sequencing, bacterial genome
This article is included in the Pathogens gateway.
This article is included in the Genomics and Genetics gateway.
subarctic soil, predatory bacteria, Myxococcota, Polyangiaceae, Mucilaginibacter cryoferens, short-read sequencing, bacterial genome
In the updated manuscript, long-read data from the same sample were added. The use of both short and long reads improved the assembly quality, making it considerably less fragmented, with the longest scaffold of 2.1 Mbp. The new assembly was also subjected to more thorough annotation, including CAZyme profiling and prediction of biosynthetic gene clusters. In addition, the updated manuscript contains new plate and microscopy images and more information about 1-FT3.2 cultivation. The new assembly was deposited in the European Nucleotide Archive (ENA) as a metagenome-assembled genome (MAG).
See the authors' detailed response to the review by Kai Blin and Judit Szenei
Predatory bacteria are important players in microbial food webs (Hungate et al. 2021). Myxobacteria are a group of bacteria associated with the phylum Myxococcota, characterised by group predatory behaviour and a complex lifestyle, where rod-shaped vegetative cells can aggregate into multicellular fruiting bodies and produce spores (Saggu et al. 2023). Myxobacteria are globally distributed and especially abundant in soil (Zhou et al. 2014; Wang et al. 2021). Together with other micropredators, myxobacteria play leading roles in carbon sequestration and mineralization in soil (Lueders et al. 2006). Moreover, myxobacteria may dominate among other potential bacterivores and have been suggested to represent one of the keystone taxa in soil microbial food webs (Petters et al. 2021). Still, more data are needed to resolve their taxonomic diversity as well as metabolic and lifestyle capacities across environments, including relatively underexplored subarctic regions.
Since soil microbial communities are highly diverse, obtaining complete genomes through metagenomics may be a challenging task (Anthony et al. 2024). Cultivating soil microbes makes it possible to reconstruct their genome sequences reliably and link genetic information to the observed phenotype. In this study, we obtained strain 1-FT3.2, a predatory bacterium from northern peatland soil in the Pallas region, Finland, using Mucilaginibacter cryoferens FT3.2 (Kumar et al. 2025) as prey. M. cryoferens, recently described as a new species, was isolated from Arctic tundra soils in the Kilpisjärvi region, Finland, where it may play important roles in litter decomposition and carbon recycling together with other Mucilaginibacter species (Männistö et al. 2009; Kumar et al. 2025). The analyses of 1-FT3.2 draft genome sequence obtained from the co-culture with its prey suggested that it belongs to the Polyangiaceae family, being only distantly related to other known representatives of this family.
A soil sample was collected from peatland in the Pallas area, Northern Finland, in September 2022 (N67°59′ E24°13′, Figure 1A). The vegetation was mainly sedges ( Figure 1B). The sample was collected from a depth of 5 cm with sterile instruments and stored at 4 °C. The pure culture of Mucilaginibacter cryoferens FT3.2 (Kumar et al. 2025), was used as the prey for isolating predatory bacteria from the soil sample. Bacteria were cultivated using R2A medium (Neogen, NCM0188A), which contained 0.5 g L −1 yeast extract, 0.5 g L −1 meat peptone, 0.5 g L −1 casamino acid, 0.5 g L −1 glucose, 0.5 g L −1 starch, 0.3 g L −1 K 2PO 4, 0.05 g L −1 MgSO 4, and 0.3 g L −1 C 3H 3NaO 3, and was adjusted to pH 6. For solid and top agar, 15 and 4 g L −1 of agar (Sigma-Aldrich, A4550) were added, respectively. The cultures were grown aerobically at room temperature (RT).

In (A), Kilpisjärvi, the original isolation location for the prey strain, Mucilaginibacter cryoferens FT3.2, is additionally shown. Map modified from Wikimedia Commons (NordNordWest). In (C), a representative plate with lysis zones on the M. cryoferens FT3.2 lawn after 14 days of incubation is shown, scale bar, 1 cm.
For the isolation, 5 g of the soil sample (wet weight) was resuspended in 50 ml of R2A broth and incubated on a shaker (~200 rpm) at RT for two weeks for the sample enrichment. The enriched sample was centrifuged (ThermoScientific F15-6x100y, 30 min, 2,500 g, 20 °C) and 100 μl of non-diluted supernatant plated with 300 μl of the M. cryoferens FT3.2 liquid culture and 3 ml of R2A soft agar (46 °C) as a top layer on R2A solid agar plates. The plates were incubated aerobically at RT. The observed growth inhibition/lysis zone was picked up with a sterile pipette tip, resuspended in R2A broth, and plated in a top agar layer as before, which was repeated three consecutive times. The strain causing lytic zones on M. cryoferens FT3.2 was named as 1-FT3.2.
For preparing 1-FT3.2 stocks, the top agar layers of the semi-confluent plates were collected and resuspended in R2A broth (3 ml per plate), incubated with shaking (~200 rpm) at RT for one hour and centrifuged (ThermoScientific F15-6x100y, 30 min, 10,000 g, 4 °C). The supernatant was collected and stored at 4 °C. The stock titers were determined by plating serial dilutions in a top agar layer as described above.
For differential interference contrast (DIC) microscopy of 1-FT3.2 cells, 1-FT3.2 stock was plated in a top agar layer as described above and incubated for three months at RT in a loosely closed box containing an open water reservoir to maintain humidity (the plates were also sealed with parafilm to prevent desiccation after the first two weeks of incubation). The colonies were collected from within the lysis zone to exclude prey cells and resuspended in R2A broth. This cell suspension was vortexed, and a 7-μl drop was placed onto a glass slide, and coverslip was applied. Images were taken with Leica DM6000B microscope at Light Microscopy Unit, Institute of Biotechnology, supported by HiLIFE and Biocenter Finland, University of Helsinki. Images were analysed with ImageJ (Fiji) (Schindelin et al. 2012).
DNA was extracted with the GeneJET Genomic DNA Purification Kit (Thermo Scientific, K0721) using the manufacturer’s protocol for Gram-negative bacteria and 20 ml of the agar stock as input. Specifically, unfiltered agar stock was centrifuged (ThermoScientific F15-6x100y, 5,000 g, 10 min, RT), and the pellet was resuspended in kit’s digestion solution. Note that the agar stocks contained cells from both M. cryoferens FT3.2 and 1-FT3.2.
For long-read sequencing, HiFi SMRTbell® Libraries were prepared using Ultra-Low DNA Input protocol. Sequencing was performed on PacBio Sequel II instrument (Pacific Biosciences, USA) and HiFi analysis in SMRTlink v12.0. For short-read sequencing, 100 ng of genomic DNA was converted to a sequencing library using the Illumina DNA prep. Samples were dual indexed using the sequencing core unit’s own Nextera primers. Seven cycles were used in the PCR step and DNA was pooled and purified using Illumina’s SPB bead purification. The Library pool was sequenced at 12 pM on the AVITI sequencer (Element Biosciences) using the AVITI 2x150 Sequencing kit Cloudbreak FreeStyle High Output. Sequencing was performed at the DNA Sequencing and Genomics Laboratory (supported by HiLIFE and Biocenter Finland funding), Institute of Biotechnology, University of Helsinki.
The quality of long reads (PacBio) was assessed with HiFiAdapterFilt v 2.0.1 (Sim et al. 2022) and they were assembled using Flye v 2.9.3 (5 iterations) (Kolmogorov et al. 2019). Here and below, contig completeness was assessed with CheckM2 v. 1.0 (Chklovski et al. 2023). The produced complete 7-Mbp contig was compared to Mucilaginibacter cryoferens FT3.2 (GenBank acc. no. NZ_CP183228.1) using BBmaps (sourceforge.net/projects/bbmap/) and pyANI v. 0.2.12 (Pritchard et al. 2015). This complete contig, which was confirmed to be the M. cryoferens FT3.2 genome, was used as a reference for read mapping with Minimap2 v.2.29 (−-secondary = yes -N 5 -p 0) (Li 2018), and unmapped reads were subjected to the assembly with flye 2.9.6 (5 iterations) and the hybrid assembly using SPAdes v. 3.15.5 (Antipov et al. 2016) together with short reads that were also filtered from the M. cryoferens FT3.2 sequences (see below).
FastQC v. 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to assess the quality of reads. Raw reads were trimmed and adaptors removed with Cutadapt v. 2.7 (−m 50 --nextseq-trim 20) (Martin 2011). Read-based taxonomic profiling was performed using phyloFlash v. 3.4.2 and SILVA138.1.eukmod database (Gruber-Vodicka et al. 2020). The full-length SSU rRNA gene sequences obtained from the phyloFlash run were searched with BLASTN (Altschul et al. 1990) against the NCBI nt database using an E-value cutoff of 0.001. Short reads were mapped to the M. cryoferens FT3.2 genome (which was obtained through the long-read sequencing, see above) using Bowtie2 v. 2.5.3 (Langmead and Salzberg 2012) and sorted with SAMtools v. 1.16.1 (−f 12 -F 256) (Danecek et al. 2021). The unmapped short reads were utilized for the hybrid assembly together with unmapped long reads using Spades v. 3.15.5 in the --isolate mode (Antipov et al. 2016). The run was repeated using contigs of ≥25 kbp in length obtained in the first hybrid assembly run as trusted contigs (−-trusted-contigs), and only scaffolds of ≥10 kbp were retained after the second run. Assembly statistics was assessed with BBTools Stats (sourceforge.net/projects/bbmap/). The obtained draft genome was classified using GTDB-Tk v. 2.3.2 with Genome Taxonomy Database (GTDB) R07-RS207 and R08-RS214 releases (Chaumeil et al. 2022) and annotated with DRAM v. 0.1.2 (Shaffer et al. 2020) and dbCAN2 search with HMMs of CAZy families v10 (E-value threshold of 1e-15) (Zhang et al. 2018) at KBase (Arkin et al. 2018). Secondary metabolite biosynthesis gene clusters were predicted with antiSMASH v 8.0.4 (detection strictness: strict) (Blin et al. 2025). Genomic distances to reference genomes in GTDB were assessed using autoMLST2.0 (Pourmohsenin et al. 2025). Putative (pro) viral sequences were predicted by geNomad v. 1.7 (Camargo et al. 2023) and their quality and completeness were assessed with CheckV v. 0.8.1 (Nayfach et al. 2021).
After about two weeks of incubating the plates inoculated with an enriched soil sample and M. cryoferens FT3.2 as prey, lysis areas of 4–5 mm were observed. In subsequent platings, the size of lytic zones reached up to ~1 cm in 14 days ( Figure 1C) and 6–7 cm in 3 months ( Figure 2A). The central parts of these zones were clear, while edges were hazier. Agar stocks produced lysis zones on the M. cryoferens FT3.2 lawn when diluted up to 10000-fold, but no lysis zones could be observed when titrating filtered stocks (0.22 and 0.45 μm PES LLG-Syringe filters Spheros), suggesting that the origin of the observed lytic zones was not viral. Very small, almost transparent or whitish colonies growing over the lysis zones were observed ( Figure 1C), growing into more visibly distinctive white colonies over time ( Figure 2A). Notably, lysis zones contained multiple colonies rather than representing single-colony plaques, suggesting collective or spreading predatory activity. However, no bigger aggregated structures like fruiting bodies were seen (at least by naked eye). An alternative cultivation approach using the myxobacterium-suited CY-C10 medium ((Karwowski et al. 1996) modified by omitting antibiotics) and higher incubation temperature (28 °C) for stock titration did not improve colony growth visibility. The tiny colonies could occasionally be subcultured to a fresh R2A plate (using a wooden stick), where the strain demonstrated prey-independent growth, although the growth remained slow and biomass production very limited ( Figure 2B). We named the strain causing lytic zones on M. cryoferens FT3.2 as 1-FT3.2.

(A) The plate with a lysis zone and visibly developed white colonies within it on the M. cryoferens FT3.2 lawn after 3 months of incubation (only one lysis zone initially observed on this plate). (B) Independently growing white colonies transferred from the plate in (A) to the R2A plate without prey supply, snapshot after one month of incubation. (C) DIC microscopy images of cells from white colonies observed in (A). Scale bar, 1 cm in (A) and (B); 10 μm in (C).
DIC microscopy of 1-FT3.2 colonies inspected after 3 months of growth at RT (supplied with prey, see Methods) revealed two types of cells: rod-shaped vegetative cell (3.22 ± 1.21 μm long, n = 27) and smaller spores (0.82 ± 0.14 μm in diameter, n = 70) ( Figure 2C). Vegetative cells were observed predominantly as aggregates.
Assembly of long-reads
Long-read sequencing with PacBio resulted in 230,474 HiFi reads (184–30,603 bp in length, 7,006 bp average length, 1.6 Gb total), none of which were adapter-contaminated. The first assembly produced one circular contig of 7,052,466 bp with the mean coverage of 226. This contig was complete (99.99% completeness, 0.9% contamination), had GC content of 42%, and shared 99.97% overall nucleotide identity and 99.99% average nucleotide identity (ANI) with the reference genome of Mucilaginibacter cryoferens FT3.2 (GenBank acc. no. NZ_CP183228.1). The strain used in this study is a laboratory-maintained derivative of the reference strain, M. cryoferens FT3.2, and the observed genome sequence difference (0.03%) likely reflects divergence arised during laboratory passage.
When long reads mapping to M. cryoferens FT3.2 genome were filtered out, 4,589 reads were retained and assembled into 161 contigs with the sum length of 5,396,049 bp (3,902–161,840 bp, 35,735 bp average length). This assembly was 61.4% complete and 0.17% contaminated and had a GC content of 64%, representing 1-FT3.2. To improve the completeness of 1-FT3.2 genome sequence, we utilized long reads that were unmapped to M. cryoferens FT3.2 together with filtered short reads for the hybrid assembly (see below).
Read-based analyses (short reads)
Short-read sequencing resulted in 245,936,278 raw read pairs (150 bp + 150 bp), of which 245,436,350 pairs were retained after read trimming and quality control. With read-based profiling by phyloFlash, 225,532 reads (0.092% of all reads) could be mapped to SSU rRNA sequences in the SILVA database. Of the mapped reads, 212,996 (94%) were assigned to the order Sphingobacteriales (Bacteroidota), where the genus Mucilaginibacter belongs to, and 9,050 (4%) were assigned to the order Polyangiales (Myxococcota). The rest of the hits constituted less than 0.01% of mapped reads each. Thus, read-based profiling confirmed two strains present in the sample, comprising ~98% of reads together. Furthermore, full-length SSU rRNA gene sequences assembled by SPAdes and matched to the SILVA database within the phyloFlash run were only two OTUs with the closest-matching references of Mucilaginibacter sp. M20–56 (Sphingobacteriales; GenBank acc. no.: KP899210.1, 99% id., 100% cov.) and Phaselicystis metagenome (Polyangiales; GenBank acc. no.: FPLS01001412.1, 95% id., 99% cov.). Additional BLASTN searches of the two detected OTUs against the NCBI nt database resulted in hits to 16S rRNA gene sequences of Mucilaginibacter sp. strain FT3.2 (100% id., 100% cov., 0 E-value) and members of the order Polyangiales (the genera Minicystis, Sorangium, Chondromyces, Labilithrix, Polyangium, and uncultured bacterium, 91–92% id., 100% cov., 0 E-value), respectively.
Hybrid assembly of 1-FT3.2 using short and long reads
When short reads mapped to the M. cryoferens FT3.2 genome that was obtained from the long-read sequencing (see above) were removed, 6,486,959 unmapped read pairs were retained for the hybrid assembly. The first run of the hybrid assembly yielded 12 scaffolds longer than 50 kb, comprising 37 contigs with 0.045% gaps. Summary statistics for the first run was the following: total length 7,644,408 bp; scaffold N50 = 1.058 Mb; scaffold L50 = 3; minimum scaffold length 52,343 bp; maximum contig length 728,505 bp; maximum scaffold length 1,629,201 bp. The second run, using trusted contigs from the first run (contigs ≥25 kb), produced 13 scaffolds >50 kb comprising 32 contigs with 0.034% gaps. Summary statistics for the second run was the following: total length 7,638,883 bp; scaffold N50 = 1.058 Mb; scaffold L50 = 3; minimum scaffold length 52,343 bp; maximum contig length 1,168,957 bp; maximum scaffold length 2,109,807 bp. Thus, the second run improved the assembly by increasing the lengths of the longest contigs and scaffolds, and this assembly (scaffolds of ≥10 kbp in length) was retained as the draft genome of 1-FT3.2 ( Table 1).
GTDB-Tk run on the draft genome suggested classifying 1-FT3.2 within the family Polyangiaceae, order Polyangiales, class Polyangia, phylum Myxococcota. With DRAM, rRNA-encoding genes were identified in the draft genome scaffolds: 16S rRNA, 5S rRNA, and 23S rRNA genes, each in five copies. The 16S rRNA gene sequence predicted by DRAM was identical to the Polyangiales OTU assembled in the phyloFlash run. With autoMLST2.0, the closest match to 1-FT3.2 was Labilithrix sp019637175 genome (GCA_019637175), sharing only 76.2% ANI. The closest match flagged as “type species” was Polyangium jinanense (GCF_028435365), having 73.7% ANI. Both matches, as well as many other top hits, belonged to the order Polyangiales.
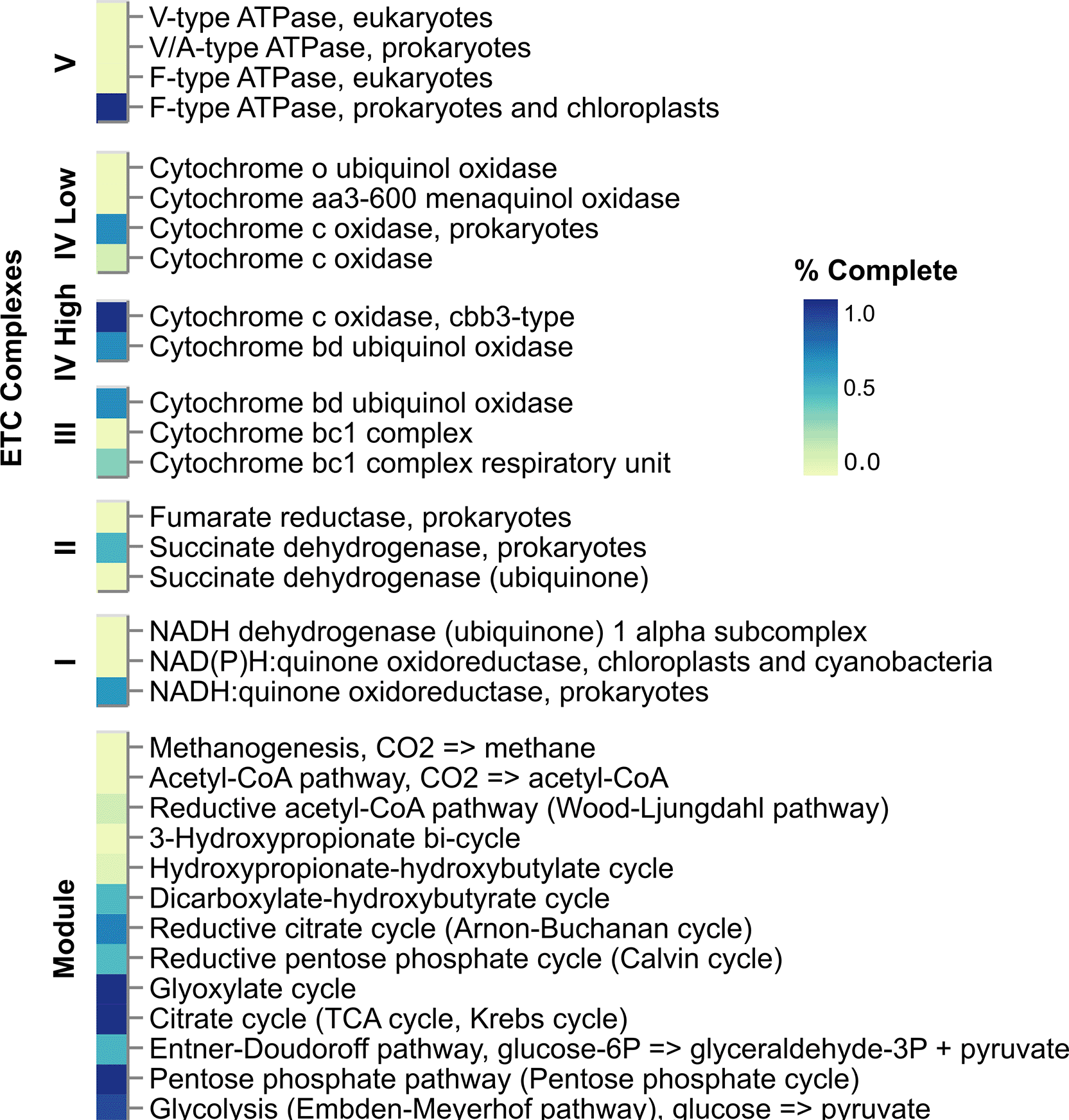
DRAM-based annotations ( Figure 3) revealed a few complete metabolic modules: pentose phosphate cycle, citrate cycle (TCA cycle), glyoxylate cycle, cytochrome c oxidase, and F-type ATPase, as well as a near-complete (8/9) glycolysis module, suggesting robust central carbon metabolism and aerobic respiration. Also, arsenate reductase (glutaredoxin), acetyl-CoA synthetase, acetate kinase, and alcohol dehydrogenase were predicted, but no CAZy enzymes. The incomplete nature of the draft genome sequence precludes full understanding of metabolic capacities or the lack of those in 1-FT3.2. Among other DRAM predictions, several different CRISPR-Cas system proteins were identified (Cas1, Cas2, Cas3, CasA, CasB, CasC, CasE, Cmr1, Cmr2, Cmr3, Cmr4, Cmr5, and Cmr6). About 36% of all predicted proteins had no significant hits to any DRAM database.
Although DRAM annotation revealed no CAZy enzymes, further search with dbCAN2 HMMs of CAZy families resulted in hits to the following enzymes: 15 families of glycoside hydrolases (GH), 10 families of glycosyltransferases (GT), four families of carbohydrate esterases (CE), two families of carbohydrate-binding modules (CBM), and two families of auxiliary activities (AA) ( Table 2). Hits the GH13 family, which comprises GHs acting on substrates containing α-glucoside linkages, included maltose alpha-D-glucosyltransferase/alpha-amylase, isoamylase, maltooligosyltrehalose trehalohydrolase, 1,4-alpha-glucan branching enzyme (also in GH57), and starch synthase (maltosyl-transferring). Other GH hits also included beta-N-acetylhexosaminidase (GH3), chitinase (GH18), soluble lytic murein transglycosylase (GH23), lysozyme (GH25), alpha-glucosidase (GH31), and 4-alpha-glucanotransferase (GH77). GTs included dolichyl-phosphate beta-glucosyltransferase (GT2_Glycos_transf_2), phosphatidyl-myo-inositol dimannoside synthase (GT4), teichuronic acid biosynthesis glycosyltransferase TuaC (GT4), starch synthase (GT5), trehalose 6-phosphate synthase/phosphatase (GT20), UDP-N-acetylglucosamine--N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase (GT28), glycogen phosphorylase (GT35), penicillin-binding proteins 1A and 1C (GT51), and arabinofuranosyltransferase (GT89). CEs were represented by S-formylglutathione hydrolase (CE1), and peptidoglycan-N-acetylglucosamine deacetylase (CE4). CBMs and Aas included expansin (CBM63) and manganese oxidase (AA1), respectively. Some hits were listed as unknown functions within families. No hits to polysaccharide lyases (PL) were retrieved.
With antiSMASH, six biosynthetic gene clusters (BGCs) were predicted in the 1-FT3.2 genome ( Table 3). These clusters included the following types (descriptions from antiSMASH glossary provided in brackets): ranthipeptide (Cys-rich peptides), hglE-KS (Heterocyst glycolipid synthase-like PKS), triceptide (Triceptides), arylpolyene (Aryl polyene), NRPS (Non-ribosomal peptide synthetase), and terpene (Terpene). From these six BGCs, only terpene showed some similarity to a known cluster (β-carotein, MIBiG acc. no. BGC0000646).
Two proviral sequences were predicted in the 1-FT3.2 draft genome at the following coordinates: 1,182,980-1,240,379 nt and 902,837–930,791 nt in the contig NODE 1_length_2109807_cov_115.129082. These proviral elements were 57,400 and 27,955 bp long and 54.5 and 74.2% complete, respectively. Both proviruses were assigned as tailed phages within the class Caudoviricetes without further classification. Although geNomad predicted a few Caudoviricetes marker genes, such as those for capsid and portal proteins, most gene predictions had no annotations in these proviruses.
A new predatory bacterium, 1-FT3.2, was isolated form subarctic peatland soil using Mucilaginibacter cryoferens FT3.2 as prey. DIC microscopy revealed cell morphologies that are typical for myxobacteria: larger rod-shaped vegetative cells (~3-μm long) and smaller coccoid spores (d ~ 0.8 μm). 1-FT3.2 is characterised by relatively slow growth and very limited prey-independent growth. A hybrid assembly of short and long sequencing reads resulted in an 82%-complete genome draft. The genome analysis placed 1-FT3.2 within the family Polyangiaceae (Myxococcota). Members of this family are terrestrial isolates mainly from soil and plant decay material, characterised by slow growth, large genomes with high GC content, and some strains being able to degrade cellulose and produce various secondary metabolites (Garcia and Müller 2014). Overall, the CAZyme profile of 1-FT3.2 suggests its ability to degrade starch and chitin, which is consistent with the characteristics reported (Garcia and Müller 2014) and predicted (Saraf and Sharma 2025) for members of Polyangiaceae. Some of the secondary metabolite types predicted in 1-FT3.2, such as terpenes, have also been reported for other Myxococcota (Saggu et al. 2023).
Polyangiaceae representatives are widespread in terrestrial environments but have rarely been isolated from subarctic soils (Dawid 2000). According to the Sandpiper database (https://sandpiper.qut.edu.au/), Polyangiaceae constitutes ~3% relative abundance in microbial communities of Arctic soils (e.g., SingleM runs for metagenomes ERR4998614, ERR4998615, ERR4998662, SRR23247776, SRR13614414, SRR13614421, SRR13614409, SRR13614411, and SRR13614413) (Woodcroft et al. 2018, 2025; Pessi et al. 2022; Bender et al. 2021), suggesting that members of this family may be more prevalent in northern soils than previously thought. The draft genome sequence of 1-FT3.2 could be used in future comparative studies aiming to resolve the diversity of the family Polyangiaceae and/or more broadly, predatory bacteria residing in subarctic soils. Based on the 16S rRNA gene sequence and whole-genome comparisons, 1-FT3.2 is only distantly related to the known species within the family Polyangiaceae and potentially represents a novel genus. Although the reported genome is incomplete, it contributes to increasing the sequenced space of the subarctic soil microbiome and the underexplored branch of Myxococcota. Having the strain available for future laboratory studies makes it possible to optimize its cultivation and explore the lifestyle and metabolic capacities in more detail. In particular, the predicted polysaccharide-degrading capacity and secondary metabolite biosynthetic potential require experimental validation.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)