Keywords
chronic migraine, Cryoneurolysis, GON
chronic migraine, Cryoneurolysis, GON
Migraine is a disabling primary headache disorder with a lifetime prevalence of 17% of the population and ranking as the second leading cause of disability in females under 50 years of age.1 Chronic migraine, defined by ≥15 headache days per month for >3 months with ≥8 migraine days, affects 1.4–2.2% of the population and imposes a major personal and socioeconomic burden.2 Current preventive treatments include repurposed oral drugs and botulinum toxin A, as well as treatments targeting the CGRP neuropeptide system, yet many patients remain insufficiently treated. Suitable treatment options for woman of childbearing potential not using highly effective contraception, pregnant and nursing woman are even more limited. Indeed, chronic migraine and related complications, such as medication overuse headache causes women of childbearing potential (WOCBP) to avoid childbearing or to having fewer children then intended,3 underscoring the need for novel non-pharmacological treatment strategies.
Greater occipital nerve (GON) block with a local anaesthetic is widely used as a transitional or rescue therapy in migraine and trigemino-autonomic cephalalgias.4 A recent randomised trial and a meta-analysis support modest efficacy in chronic migraine, with reductions in headache days and responder rates superior to placebo.5 However, short duration of effect and the need for repeated injections limit its preventive utility.
The analgesic effects of GON blocks on headache disorders, often characterized by pain in trigeminal regions, suggest a functional interaction between the occipital and trigeminal systems. The functional overlap between occipital and trigeminal sensory processing takes place at the level of the upper cervical spinal cord where nociceptive fibres from both regions converge on the trigeminocervical complex ( Figure 1). This functional and anatomical coupling is demonstrated in several animal studies.6–10 In humans, when the greater occipital nerve (GON) is pharmacologically blocked in participants without a headache diagnosis or other pain condition, a reduced nociceptive trigeminal response was observed compared to placebo.11 Similar studies have not been performed in headache sufferers.
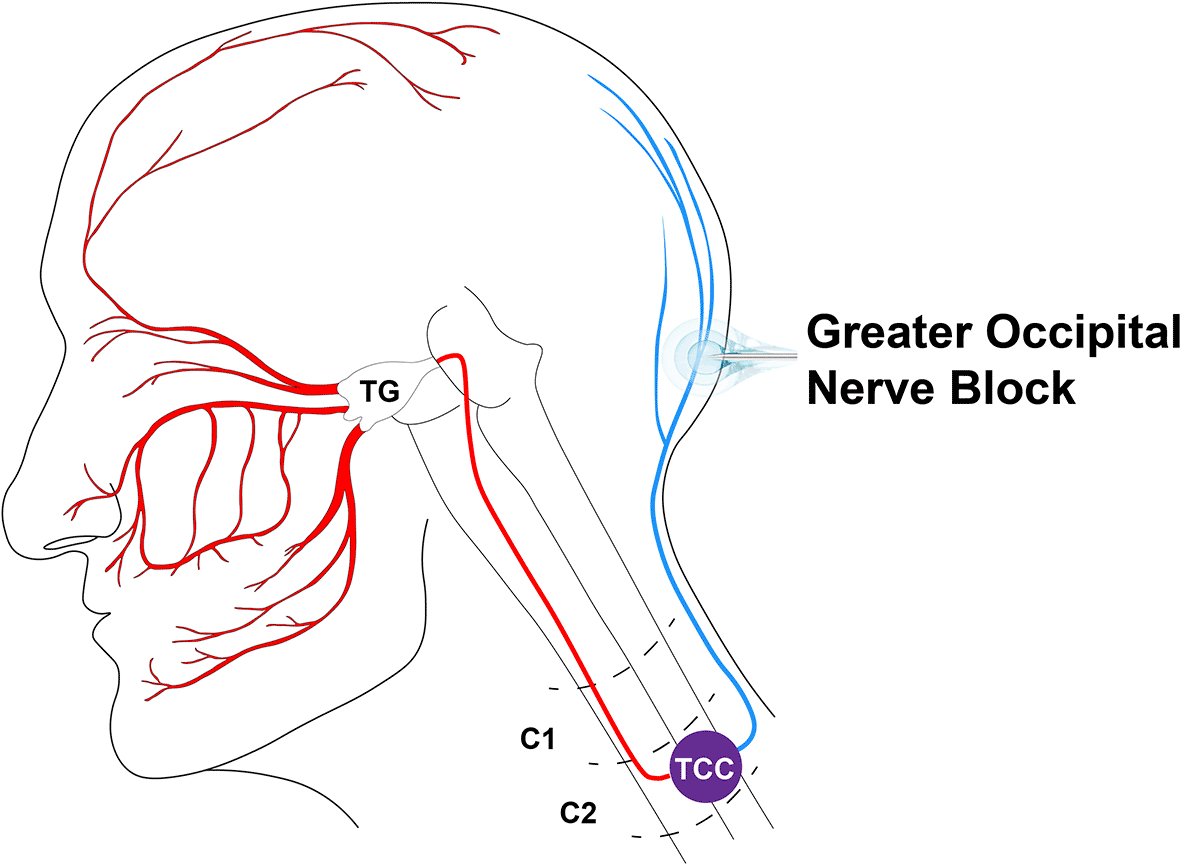
Together, these nerves provide sensory innervation to the entire head and are functionally coupled within the trigeminocervical complex (TCC). A needle illustration indicates the approximate segment of the greater occipital nerve targeted by conventional GON blocks and by the CryoGON procedure. Bilateral treatment (not shown) is performed for both interventions.
Cryoneurolysis is an established treatment in pain medicine.12 By applying cold temperatures to neural structures, it induces reversible Wallerian degeneration (axonotmesis) with subsequent regeneration, resulting in a long-acting but reversible nerve block. Cryoneurolysis has demonstrated efficacy in trigeminal and occipital neuralgia, with reported pain relief lasting several months.13–16 A sham-controlled study in cervicogenic headache, in which the control-arm received GON-block with steroids, did not demonstrate a statistically significant between-group effect. The investigators in this trial used anatomical landmarks and nerve stimulation to guide the cryoprobe, and insufficient targeting could have reduced the efficacy in the cryoneurolysis arm. It is to date no published reports on efficacy or safety in primary headache conditions to our knowledge, but cryoneurolysis towards the GON is currently studied in an open label pilot study in chronic migraine (NCT06554886).
Surgical navigation systems enable accurate percutaneous targeting of deep neural structures using pre-procedural imaging and real-time tracking. Applied to the GON, navigation-guided cryoneurolysis (CryoGON) offers a minimally invasive, office-based procedure with the potential to provide longer-lasting preventive benefit than conventional GON blocks.
This trial is designed to determine whether response to a conventional GON block predicts subsequent response to CryoGON in chronic migraine, thereby assessing the clinical utility of the block as a decision tool. Secondary objectives include evaluation of open-label efficacy, safety, tolerability, and feasibility of the CryoGON procedure. To that end, this trial will inform the design of future studies by assessing the feasibility and rational for conducting of a sham-controlled trial.
Primary objective
To evaluate whether clinical response to a conventional greater occipital nerve (GON) block predicts treatment response to navigation-guided cryoneurolysis of the GON (CryoGON) in patients with chronic migraine.
Secondary objective
To evaluate the open label clinical performance of CryoGON as a potential new treatment for chronic migraine.
The study is planned to be initiated early 2026 with a duration of recruitment for 2 years ending early 2028. Data lock is thus planned mid-2028 and submission of the stage 2 manuscript by end of 2028. Publication will adhere to the Vancouver guidelines. We do not intend to use professional writers.
This protocol publication is based on the clinical investigation plan (study protocol), version 1.0, dated 14th of October 2025. This will be a clinical investigation of a medical device for cryoneurolysis, that will be conducted in accordance with the Declaration of Helsinki, Norwegian law (including the Medical Device Regulation), and ISO 14155:2020 (Clinical investigation of medical devices for human subjects – Good clinical practice). The trial will be initiated following approval by the competent authorities, the Regional Committee for Medical and Health Research Ethics and institutional approval by the sponsor. The inclusion and data collection of each study participant will commence only after their written informed consent has been obtained. Amendments to the protocol will be implemented only after appropriate review and approval by the relevant parties. The study will be prospectively registered at ClinicalTrials.gov prior to enrolment of the first participant. All items from the WHO Trial Registration Data Set will be included in the ClinicalTrials.gov record and correspond to the information provided in this protocol publication.
The study is investigator-initiated and will be sponsored by a public hospital (St Olav’s university hospital, 7006 Trondheim, Norway, [email protected]). It will be conducted under the auspices of the Norwegian Headache Research Center (NorHead), a publicly funded academic center at the Norwegian University of Science and Technology. Funding for this study will be provided by the DAM Foundation (Stiftelsen Dam, grant no SDAM_FOR701626), a Norwegian non-profit foundation that allocates research grants from the surplus of the national lottery operator (Norsk Tipping). The grant application was submitted in collaboration with the national patient organization Hodepine Norge (eng. Headache Norway), which also coordinates patient involvement in the design and conduct of the study. A prerequisite of the DAM Foundation grant award is that dissemination follows the two-stage Registered Report publication format.
The funding body, sponsor or any other party than the authors of this paper, had no role in the study design, in writing the clinical investigation plan (study protocol) and the decision to submit the protocol for publication.
Participants are insured through the Norwegian System of Patient Injury Compensation (Norsk pasientskadeerstatning). No post-trial care is provided with-in the framework of the trial.
The trial will be conducted as a single-site, open-label study within the Norwegian public healthcare system, which reimburses patient travel and accommodation expenses. Should any co-payment be required, these costs will be covered by the study. No remuneration or financial incentives will be offered beyond reimbursement of necessary expenses. The study will be designed and conducted according to the guidelines of the International Headache Society.17
Study recruitment
Recruitment will be facilitated through the NorHead national research infrastructure. Once enrolment opens, study information will be made available on governmental and institutional websites as well as through social media platforms. Patients may be referred by their treating physician or may self-register via the NorHead online portal. Study personell will perform pre-screening of candidates, and only patients considered potentially eligible will be scheduled for a formal eligibility assessment by a neurologist.
Eligibility criteria
Inclusion and exclusion criteria are summarized in Table 1. In brief, eligible participants will be adults (≥18 years) with chronic migraine, with or without aura, fulfilling the International Classification of Headache Disorders (ICHD-3) criteria and verified by a neurologist at inclusion. Participants must have a history of chronic migraine for at least one year, with onset of episodic migraine before age 50 and onset of chronic migraine before age 65. Preventive medications must have been stable for at least three months (or ≥5 half-lives, whichever is longer) prior to screening, and participants must agree to maintain their regimen throughout the study. Individuals treated with onabotulinumtoxinA must have received at least three consecutive PREEMPT-protocol injection cycles with stable intervals. Only patients deemed suitable for both a conventional GON block and CryoGON by the investigator, and capable of providing written informed consent, will be enrolled.
Key exclusion criteria include prior GON-targeted injections within 12 weeks of baseline, inability to differentiate migraine days from other headache types, or coexisting secondary headache disorders (except medication overuse headache). Patients with occipital neuralgia or neuropathy or other pain conditions that may confound evaluation of study outcomes will be excluded. Additional exclusions include unstable preventive medication regimens during baseline, prior or current occipital nerve stimulation, frequent opioid or any barbiturate use, significant psychiatric comorbidity or abnormal pain behaviour, relevant medical conditions increasing procedural risk (e.g. bleeding diathesis), and ongoing treatment site infection.
Study outline
The study will comprise four in-clinic visits and a series of structured follow-up contacts, separated by observation periods with prospective daily eDiary data collection (Figure 2). At visit 1, eligibility will be assessed and written informed consent obtained via an electronic signature in a secure institutional digital platform (the eFORSK platform). Eligible participants will then enter a minimum 28-day baseline period, which serves both as a screening phase and to establish baseline data for endpoint assessments. A diary adherence of at least 90% (≥25 of 28 days) will be required to proceed. At visit 2, participants will be re-screened, and those meeting criteria will receive bilateral conventional greater occipital nerve (GON) blocks followed by a minimum 28-day observation period. A structured telephone call will be conducted one week after the procedure to capture any treatment-emergent adverse events and confirm scheduling for visit 3. At visit 3, eligibility will again be reassessed, including review of contraindications to CryoGON, confirmation of ≥90% diary adherence, and stable prophylactic regimens. Participants meeting criteria will undergo the CryoGON procedure (navigation-guided cryoneurolysis of the GON). Structured telephone follow-ups will be performed at weeks 1, 4, and 8, and a final in-clinic assessment will be conducted at week 12 post-CryoGON.
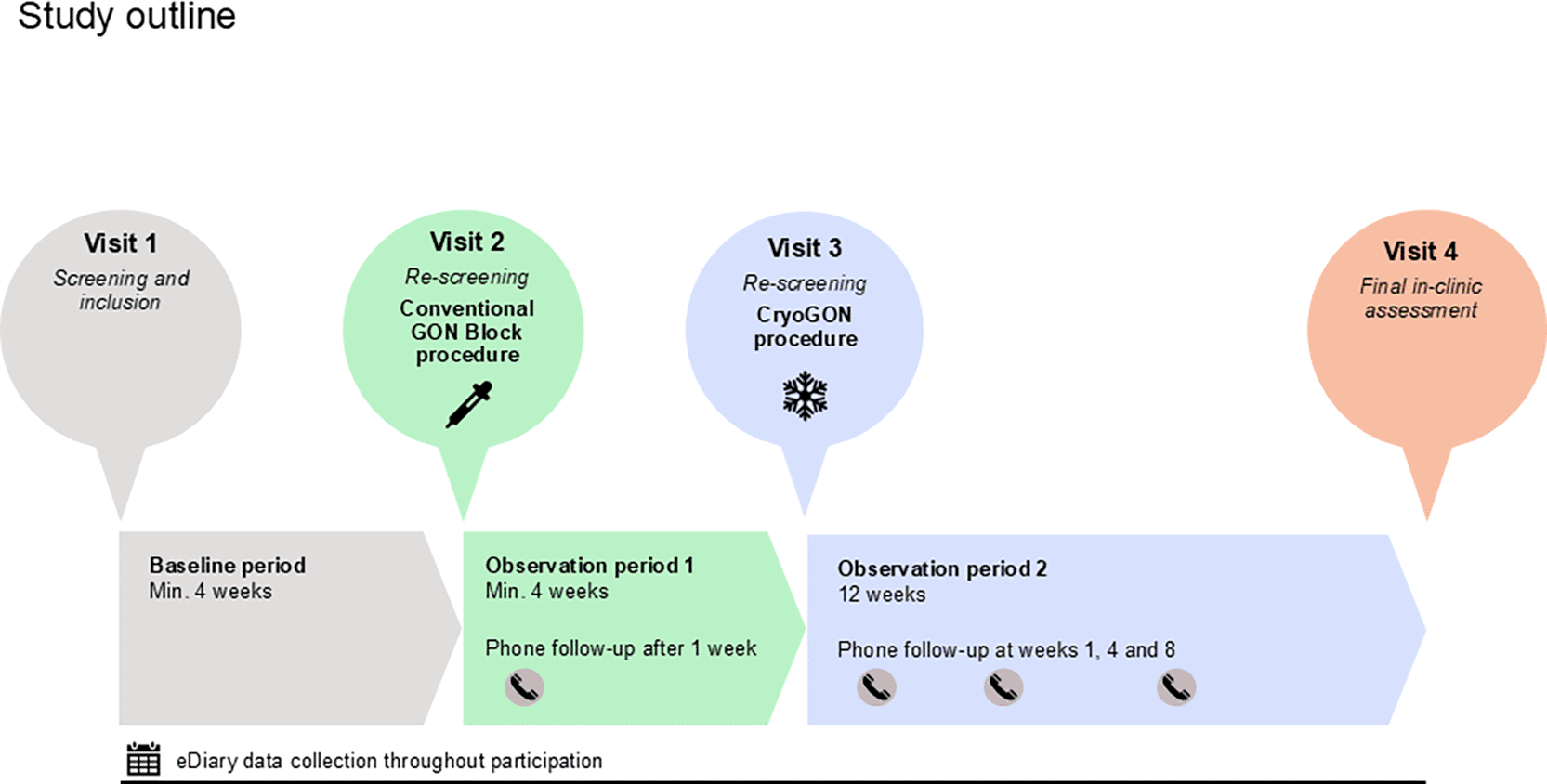
The study includes four in-clinic visits separated by observation periods with daily prospective eDiary recording.
Considerations for woman of childbearing potential
This study will include women of childbearing potential irrespective of contraceptive use, as well as pregnant and breastfeeding women. In accordance with Article 66 of the Medical Device Regulation (MDR 2017/745), pregnant and breastfeeding participants require additional protection, including specific justification and risk minimisation measures where appropriate.
In chronic migraine, only CGRP-targeting treatments, topiramate, and onabotulinumtoxinA are supported by high-quality clinical evidence. Topiramate has recently fallen out of favour for women of childbearing potential due to the risk of neurodevelopmental disorders [12], while the safety of onabotulinumtoxinA and CGRP-inhibitors during pregnancy remains unknown, rendering them contraindicated. Consequently, effective prophylactic options are highly limited during the period when women are trying to conceive and throughout pregnancy.
Local anaesthesia administered towards the greater occipital nerve (GON) either as part of the conventional GON block or as anaesthesia as part of the CryoGON procedure, is not contraindicated in these populations, and conventional GON blocks are already in clinical use in pregnant and breastfeeding patients.18–20 Cryoneurolysis is a non-pharmacological treatment, not contraindicated in these populations, and there is no plausible rationale to suspect increased risk of harm to a nursing child or an embryo or fetus. Given the very limited alternative treatment options available, inclusion of pregnant and breastfeeding participants in this study is considered acceptable.
Conventional GON-block
The conventional GON block will be performed bilaterally in an outpatient setting by trained investigators. The injection site is identified at the medial third of a line drawn between the external occipital protuberance and the mastoid process. After sterile skin preparation, a 23-gauge needle attached to a 5 mL syringe is inserted perpendicular to the skin until firm resistance is encountered, indicating contact with the periosteum. The needle is then withdrawn slightly, aspiration is performed to exclude arterial entry, and the needle is redirected superiorly.
A total of 4 mL per side of a 50:50 mixture of lidocaine 2% and bupivacaine 0.5% is injected slowly in a fan-shaped distribution to ensure adequate spread. The procedure is repeated contralaterally.
The CryoGON procedure will be performed as an image-guided, percutaneous intervention. Image modalities may be per-procedural ultrasound guidance and/or surgical navigation based on MRI neurography. Devices will be used in accordance with their intended purpose and instructions for use (IFU). Study interventionists are trained surgeons with prior experience using navigation and cryoneurolysis technology and will perform the procedure according to the IFU.
For each procedure, local anesthetics will first be administered with 2–8 mL of lidocaine/adrenaline. A small skin incision of approximately 2 mm is then made at the predetermined entry point near the GON. The cryoprobe is advanced towards the target nerve under image guidance using the navigation system. Once appropriate positioning is confirmed, cryoneurolysis is applied in three cycles of 1 minute each, after which the probe is carefully retracted.
Participants will be observed 30 minutes post-procedure to monitor for acute adverse events and discharged home thereafter.
Clinical experience spanning nearly half a century supports the safety of cryoneurolysis as a therapeutic approach in pain management.21–23 Known adverse device effects include injection site bleeding, hematoma and bruising. Injection site infection is reported as very rare.24,25 In contrast to other neuroablative methods, cryoneurolysis has demonstrated a low risk for neuritis, and if it occurs, it is transient. However, transient dysesthesia of a severity that affected activities of daily living has been reported, occurring in 1,2% in one study.26 Long term neuropathy induced by cryoneurolysis has never been reported.21,23 Treatment of superficial nerves can cause hyperpigmentation, depigmentation and alopecia at the treatment site, and excessively superficial lesioning can cause skin frostbite.23 In five studies that targeted the occipital nerve,14–16,27,28 reporting from a total of 172 participants, mostly minor and transient adverse events occurred. One hematoma and 2 postprocedural occipital neurits were also reported. Table 2 presents a list of known or anticipated adverse device effects associated with cryoneurolysis.
MedDRA = Medical dictionary for regulatory activities, TEAE = Treatment emergent adverse events.
A predefined source data list will specify what constitutes source data in this investigation. Source documentation includes electronic patient records (EPJ) for medical history, eligibility assessments, medication use (including preventive treatments), headache diagnoses, and safety data. Data handling will comply with the General Data Protection Regulation (GDPR) and institutional requirements. Study data will be retained for 25 years after completion, as mandated by Norwegian law. Data management will follow a study-specific data management plan.
eCRF
An electronic Case Report Form (eCRF) will be implemented using the VieDoc platform (Viedoc Technologies AB), supported via the research department at St. Olav’s University Hospital. Information obtained during visits, telephone consultations, and all safety events will be entered into the eCRF. Data are stored in a pseudo-anonymised format, where participants are identified only through a unique study identification number. In case of active withdrawal, the reason (if any) will be registered in the eCRF.
eDiary
The eDiary application serves as the primary tool for endpoint data collection. Participants will complete daily entries throughout the baseline, GON-block, and CryoGON follow-up periods, regardless of headache status. On headache days, participants report maximum pain intensity, duration, the presence of aura, abortive medication use, and whether the headache is a migraine. A section to report alteration in participant health (e.g. potential adverse events) is available every day.
Investigators access an online dashboard that enables monitoring of diary adherence and identification of participants with insufficient entries. Entries can be made retrospectively for up to three days (current day plus the two preceding days).
Safety data
Safety data are collected through structured interviews at visits and telephone follow-ups, and in case of participants making contact in-between. The online dashboard presents patient-reported potential adverse events from the eDiary, which will be reviewed during study contacts. All treatment-emergent adverse events (AEs) will be registered in the eCRF.
Questionnaires
The Migraine-Specific Quality of Life Questionnaire v2.1(MSQ) and the Patients’ global impression of change (PGI-C) will be administered to participants. The MSQ will be collected at baseline and at week 12 following CryoGON. The PGI-C will be collected at week 1 and 4 following conventional GON and week 8 and 12 following the CryoGON-procedure. All copyrighted instruments will be used with proper permission or license.
Endpoint variable definitions follow the International Headache Society (IHS) guideline for controlled trials of preventive treatment of chronic migraine17 (Table 3). The primary endpoint variable for this study is moderate to severe headache days. Several endpoints (Table 4) are predefined to address the two main objectives of the study, i.e. assessing whether short-term response to a conventional GON block predicts subsequent response to CryoGON and evaluating the clinical performance of the CryoGON procedure.
To address the primary objective, the primary endpoint is the change in moderate–severe headache days from baseline to weeks 9–12 after CryoGON, analysed as a continuous outcome. The primary analysis estimates the association between change after the GON block (weeks 1–4; primary predictor) and change after CryoGON (weeks 9–12).
While the primary endpoint aims to evaluate whether there is an association between degree of response to conventional GON block and CryoGON treatment, this does not necessarily translate to clinical utility. A statistically significant correlation may indicate that patients who benefit more from the GON block also benefit more from CryoGON, but may not justify the use of GON block response as a binary decision-making tool.
To that end, we will estimate the predictive capability of GON block response in determining which participants will respond to CryoGON. In this analysis we will identify clinical responders, defined as a ≥30% reduction in moderate–severe headache days from baseline, in both weeks 1-4 after GON block and weeks 9–12 after CryoGON. The proportion of CryoGON responders will be compared between those who did and did not respond to GON block, estimated with a risk ratio and 95% confidence interval (CI). Additionally, the sensitivity, specificity and predictive values of response/non-response to GON block will be estimated to identify the usefulness of this treatment as a diagnostic test for response to CryoGON. These measures will also be presented with 95% CI. The 30% responder threshold is selected as it is reported to translate to a clinical meaningful change in chronic migraine.29 Two additional analyses with different responder thresholds definitions are also prespecified, namely (i) 50% responder threshold and (ii) a patient-reported impression of change threshold, based on improvement on PGI-C collected at the end of study participation.
Open-label efficacy endpoints
The open-label efficacy of CryoGON will be evaluated based on patient-reported prospective headache diary outcomes and quality of life and patient global impression questionnaires. The migraine day endpoint variable definition is operationalized as any day with headache of minimum 4 hours duration that the participant self-identifies as a migraine, or any headache day that is successfully treated with the use of triptans and/or gepants. Patients unable to distinguish a migraine day from other headache semiologies are ineligible according to study criteria. As such, the ability of participant to identify a migraine day is part of the review at the time of inclusion and ensures that the endpoint is operationalized in a manner that adheres to the HIS’ guideline.17 Change in quality of life will be measured with the MSQ v2.1 questionnaire at baseline and week 12. Patient global impression of change will be collected at week 8 and week 12.
Open label safety, tolerability, and feasibility endpoints
Safety will be assessed separately for CryoGON (weeks 1-12 following the CryoGON procedure) and conventional GON block (weeks 1-4 following conventional GON injection) as treatment-emergent adverse events (TEAEs) and treatment emergent serious adverse events (TESAEs), adverse device effects (ADEs) and serious ADE (SADEs). Treatment-emergent refers to all AEs events at or following initiation of the conventional GON (week 1-4) or CryoGON procedure (week 1-12), respectively. All AEs will be coded according to the Medical dictionary for regulatory activities (MedDRA) and grouped and summarised using the lowest level term (LLTs).
Severity assessment will apply the standardized grading criteria form the Common Terminology Criteria for Adverse Events (CTCAE) version 6. Casualty assessment will follow the definition of the Medical Device Coordination Group,30 and all TEAE will be categorized as not related or possible, probable or casual related to conventional GON in week 1-4 post-procedure and the CryoGON-procedure in week 1-12 post-procedure. An adverse device effects (ADE) is defined according to ISO 14155:2020 as any adverse event related to the use of an investigational device. This is operationalised for this study as any AE that is assessed to have possible, probable or causal relationship to the CryoGON procedure
Tolerability will primarily be evaluated by intra-procedural discomfort (NRS 0–10) and additionally, the willingness of the participant to repeat the CryoGON procedure. At week 12, or earlier in the event of participant withdrawal, participants will be explicitly asked to consider the balance of perceived benefits and drawbacks of CryoGON and respond with ‘yes’, ‘no’, or ‘unsure’ regarding willingness to undergo the procedure again. The primary feasibility endpoint will be technical success rate. Technical success is defined as a composite of (i) surgeon-confirmed completion of the planned cryo-lesion at the intended target without device deficiency, and (ii) participant-reported numbness persisting >72 hours in the GON peripheral innervation area as reported by the participant on a telephone call after one week. The second criterium is included with the intention to distinguish prolonged sensory change consistent with cryoneurolysis from transient anaesthesia due to local anaesthetic used during the procedure. Other feasibility endpoints are number of device deficiencies, CryoGON procedure duration and diary adherence across study periods.
Adequate diary adherence is essential for the valid estimation of headache day–based endpoints, particularly responder rates. Therefore, diary adherence is included as an operational feasibility endpoint to enable evaluation of the robustness of the data underpinning both the primary endpoint and the open-label efficacy analyses.
This trial is designed to evaluate if and to what degree response to conventional GON blocks predict response to CryoGON-treatment. For the primary endpoint the following hypothesis is testet:
• Null hypothesis (H0): There is no relationship between the degree of treatment response to conventional GON block and CryoGON treatment (regression slope (β1) = 0)
• Alternative hypothesis (H1): There is an association between the degree of treatment response to conventional GON block and CryoGON treatment (regression slope (β1) ≠ 0)
Protocol violations in this study are headache diary nonadherence defined as <90% adherence and/or alterations in headache preventive treatment during trial participation. Electronic headache diary adherence will be calculated at 4-week (28 days) intervals.
The main analysis is planned when all participants have concluded the 12-week follow-up after the CryoGON procedure and the database has been locked. A statistical analysis plan (SAP) will be finalized prior to data lock.
The full analysis set (FAS) will include all participants that received CryoGON-treatment to adhere to the intention to treat ideal and will be used for the main analysis. Two additional populations are defined for the purpose of sensitivity analyses. The per protocol set (PPS) includes participants without protocol violations. A technical success set (TSS) includes the subset of participants that fulfil the criteria for a technical successful CryoGON-treatment.
The analysis of the primary endpoint will use linear regression, with change after CryoGON as the dependent variable and change after the GON block (measured during weeks 1–4) as the main predictor. For participants who do not complete the eDiary on all days, the number of moderate-severe headache days will be calculated based on the available data and estimated for a 28-day period before calculating the change in number of moderate-severe headache days.
The key secondary outcome is defined as a ≥30% reduction in moderate to severe headache days from baseline to weeks 9–12 after CryoGON, treated as a binary response (yes/no). The corresponding GON block response is also defined dichotomously using the same threshold, assessed during weeks 1–4. The proportion of participants with ≥30% response to CryoGON will be compared between those who did and did not respond to GON block using a risk ratio with 95% confidence interval (CI).
Additional diagnostic performance measures—sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV)—will also be calculated with 95% CI.
As this is an exploratory pilot study and the study has one specified primary endpoint, no adjustment of multiplicity is planned.
We prespecify a key sensitivity analysis restricted to the TSS population to estimate effect sizes conditional on technical successful CryoGON delivery as per prespecified criteria. We highlight this analysis because it reflects the expected performance of CryoGON when delivered as intended and might better mirror real world practice, where an unsuccessful first attempt would typically prompt retreatment rather than be taken as evidence of inefficacy. To preserve interpretability and precision of this key sensitivity analysis, recruitment will continue until the TSS population reaches the sample size specified by the power calculation.
Additional sensitivity analysis
• Restricted analysis to the per protocol population.
• Adjusted analysis to account for cyclical variation in headache frequency related to injection-based preventives (e.g., onabotulinumtoxinA, CGRP monoclonal antibodies). Analysis adjusting for other variables, such as treatment expectation, sex and age will be prespecified in the SAP.
Subgroup analyses will be prespecified in the SAP, and may include exploring differences in effect according to the degree of numbness following CryoGON (hypoaesthesia vs anaesthesia) and comparing bilateral versus unilateral successful CryoGON treatment.
The primary scientific question is whether the response to the GON block can predict treatment outcome after CryoGON. This will be assessed both in terms of whether there is a linear association between the degree of response to GON block and response to CryoGON (primary endpoint), and whether a ≥30% response to GON block is associated a greater chance of response to CryoGON and can be used as a diagnostic test to identify those who are likely to respond to CryoGON (key secondary endpoints). Both analysis strategies are considered in the sample size calculation, together with an assessment of what are feasible recruitment numbers. Ultimately, the target samples size was set to 70 based on feasibility and the importance of the precision of estimates.
The primary analysis tests the association between change in moderate-to-severe headache days after the GON block (weeks 1–4) and change after CryoGON (weeks 9–12) using linear regression (with the null hypothesis that there is no association between the response to GON block and CryoGON, i.e. β1 = 0). The proposed advantage of CryoGON is a much longer duration of treatment effect than is observed with conventional GON block. Thus, an association of 0.5 will be considered clinically meaningful, that is to say the gradient of the regression slope is 0.5, and thus for each 1-day reduction in moderate-severe headache days in weeks 1-4 after GON block, the average reduction is half a day in week 9-12 following the CryoGON-procedure. Assuming a standard deviation 5 for change in number of moderate-severe headache days from baseline, 26 participants will be sufficient to provide 80% power to detect a regression slope of 0.5 at 5% significance level.
The key secondary outcomes include a comparison of the proportion of responders to CryoGON (≥30% reduction in moderate-severe headache days) among those with and without response to GON block. This will be reported as a risk ratio (95% CI), along with sensitivity, specificity, PPV and NPV (also with 95% CI). We expect that around 40% will respond to GON block based on results from Chowdhury et al. (2021),5 who reported ~20% response using a 50% threshold. Among those who do not respond to GON block, we anticipate that around 30% will respond to CryoGON. For GON block to provide reasonable predictive information, the response to CryoGON among those who respond to GON block should be around 80% (i.e. a 50-percentage point increase in response). This would also equate to a sensitivity of 64% and specificity of 80%. A sample size of 31 (18 in the GON block negative and 13 in the GON block positive group) will be sufficient to achieve 80% power to detect a 50-percentage point difference (and risk ratio of 2.67) at 5% significance level and using a Chi-squared test. However, this would provide imprecise estimates of the sensitivity and specificity of GON block. With a target sample size of 70, we will have sufficient data to estimate a 95% CI around the sensitivity and specificity of a width of approximately 17 and 12 percentage points, respectively.
The CryoGON trial will be monitored by independent clinical research monitors in accordance with a dedicated monitoring plan. The plan will be risk-adapted and developed on the basis of a project-specific risk analysis. Monitoring will focus on data integrity, adherence to protocol, regulatory compliance, and participant safety.
An independent Data Safety Monitoring Committee (DSMC) will be established. The DSMC will review accumulating safety data after approximately 25% and 50% of participants have undergone the CryoGON procedure, and will provide recommendations to the sponsor regarding continuation, modification, or termination of the trial based on the observed safety profile.
At the time of database lock following study completion, only the study statistician will be granted full access to the dataset for the purpose of conducting the analyses prespecified in the Statistical Analysis Plan (SAP). After the preplanned analyses are finalised, the full pseudonymised dataset will be made available to all investigators involved in the study, enabling independent verification of results and preparation of publications. Data sharing will be governed by applicable data protection legislation and no contractual restrictions will apply.
The present study protocol outlines a prospective clinical investigation of image-guided cryoneurolysis of the greater occipital nerve (CryoGON) for the preventive treatment of chronic migraine. Conventional local anaesthetic GON blocks are already in use for transitional or acute treatment in headache care, but their short duration of effect limits their role as a preventive strategy. Cryoneurolysis, by inducing a reversible long-acting nerve block through controlled axonotmesis, offers the possibility of a more sustained relief. If effective, this approach may open an entirely new non-pharmacological treatment avenue for chronic migraine, a patient group often in need of additional options.
This trial makes two key contributions. First, it will generate the largest open-label dataset to date on safety, tolerability, feasibility, and efficacy of CryoGON in chronic migraine. These data will inform the design of future studies, including the feasibility and justification of a sham-controlled trial. Second, it will evaluate the predictive value of a conventional GON block in identifying likely CryoGON responders. The primary endpoint assesses the continuous relationship between reduction in headache days following the GON block and after CryoGON, thereby addressing the scientific question of whether the two responses are correlated. The key secondary endpoint complements this by estimating the diagnostic performance of a dichotomised response threshold (≥30% reduction), reporting sensitivity, specificity, positive and negative predictive values. Together, these endpoints provide clinically applicable evidence that can suggest whether GON block response can serve as a decision-making tool for selecting patients for CryoGON treatment.
The study is conducted within a research environment with long-standing expertise in investigator-initiated interventional trials in headache disorders. The infrastructure for recruitment, eDiary-based outcome collection, and rigorous monitoring ensures both high level of data quality and study feasibility. User involvement has been central from the outset. Patient representatives from the Norwegian patient organization contributed to the decision to prioritise chronic migraine for this trial and remain engaged in its planning and implementation. This aligns the study with patient priorities and strengthens its potential for clinical translation.
This project contains the following extended data: (Wergeland, Tore (2025))
Spirit cheklist: https://doi.org/10.6084/m9.figshare.30355048.31
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)