Keywords
Aquaculture, Nanopore, Illumina, Arabian Gulf, teleost, Serranidae, Qatar, whole genome sequencing
This article is included in the Genomics and Genetics gateway.
Aquaculture, Nanopore, Illumina, Arabian Gulf, teleost, Serranidae, Qatar, whole genome sequencing
Epinephelus coioides (Hamilton, 1822), commonly known as the orange-spotted grouper or hamour in the Arabian Gulf region, is a fish species that belongs to the family Serranidae (Rimmer and Glamuzina, 2019). It is widely distributed across the Indo-Pacific region, including the coastal waters of the Arabian Gulf, comprising Qatar’s coastal regions. This species is highly valued for its meat quality, is a popular target for both commercial and recreational fishing, and is widely cultured around the world particularly in the Asia-Pacific region (Ranjan et al., 2017). While hatchery production has been increasing to mitigate the pressures on wild populations (Antoro et al., 2006), juveniles are still being captured from the wild for mariculture operations (Tupper and Sheriff, 2008), with potential impacts for wild population sustainability.
The hamour presents a distinctive appearance with an elongated body and a large mouth. Its coloration can vary, but it typically features a brownish or reddish background adorned with prominent orange or reddish spots, giving it its common name. It has a robust build and can reach large sizes, with adult specimens sometimes exceeding one meter in length (Chen et al., 2005). This species constitutes a highly valued commodity, reaching substantial prices in the international market, for example ranging from 11.70 to 40.30 USD per kilogram with an average of 21.45 USD per kilogram in Hong Kong and south east Asia (Ranjan et al., 2017). In the Arabian Gulf, it is the most important reef-associated commercial species, collected via demersal traps, longlines, and trawls (Grandcourt et al., 2009). In northern Oman and Iran, it is targeted using wire basket traps and is also commonly taken as bycatch in trawl fisheries (McIlwain et al., 2016).
Genome sequencing plays a vital role in understanding the genetic makeup of a species, including both genes and regulatory elements, and facilitates understanding of its evolutionary history. By analysing its genome, insights into multiple aspects of a species’ biology, such as disease resistance, growth patterns, and reproductive characteristics can be obtained. To date, several genomics studies have been performed in this species, for example, genome-wide association studies were used to explore ammonia tolerance (Xu et al., 2019), a highly flexible and repeatable single nucleotide polymorphism genotyping method was employed to study its growth and ammonia tolerance (Shan et al., 2023), and whole genome sequencing and analysis revealed key regulatory pathways influencing sex differentiation (Li et al., 2023).
Here, we present a highly continuous chromosome-level genome assembly of this species, obtained using long and short read sequencing technologies and Omni-C scaffolding.
One individual male hamour (43 cm) was caught from the wild (East Qatar, Arabian Gulf, September 2022), and sampled directly. For genomic DNA extraction, 50 mg of liver tissue were excised, submerged in isopentane bath (prepared over dry ice) until frozen (30 s), stored in a pre-chilled cryotube and preserved at -80 °C until further analyses. In parallel, 50 mg of tissue from liver, kidney, heart, spleen, gonad, muscle, skin, gill, and tail were collected from the same specimen and placed in cryotubes containing RNAlater® (Sigma-Aldrich), stored at +4 °C for 24 hrs to ensure appropriate tissue preservation, and then moved to -80 °C until RNA extraction.
High molecular weight (HMW) DNA was extracted from the liver sample using QIAGEN® Genomic tip Blood & Cell Culture DNA Midi Kit (QIAGEN) following the manufacturer’s protocol. Purified DNA was quantified using an Invitrogen™ Qubit™ DNA BR Assay kit (Thermo Fisher Scientific), the purity of the sample was analysed using a NanoDrop™ 2000 spectrophotometer (Thermo Fisher Scientific) UV/Vis, and the integrity of the DNA was assessed with Femto Pulse Genomic DNA 165 kb kit (Agilent). DNA was stored at +4 °C until library preparation.
Total RNA was extracted using a Promega Maxwell® RSC 48 Instrument and Maxwell RSC simply RNA Tissue kit (Promega), quantified using an Invitrogen Qubit RNA BR Assay kit (Thermo Fisher Scientific), and analysed for purity using an RNA 6000 Nano Bioanalyzer 2100 Assay (Agilent). Purified RNA samples were stored at -80 °C until library preparation.
A long-read library for Oxford Nanopore Technologies™ (ONT) sequencing was prepared from DNA extracted from the liver using the 1D Sequencing kit SQK-LSK110 (ONT). In brief, 3 μg of DNA underwent end-repair and adenylation using the NEBNext® Ultra™ II End Repair/dA-Tailing Module (New England Biolabs), followed by ligation of sequencing adaptors. The ligation product was purified using Beckman Coulter™ AMPure XP Beads (Beckman Coulter Life Science) and eluted in Elution Buffer (ONT). The library was sequenced on a PromethION™ 24 instrument with a R9.4.1 flow cell, and data collected for 110 hours. The quality parameters of the sequencing run were monitored in real time using the MinKNOW™ platform v22.10.7 (ONT, https://nanoporetech.com/document/experiment-companion-minknow) and basecalling performed using Guppy v6.3.9 (ONT, https://nanoporetech.com/document/Guppy-protocol ).
For the proximity ligation library, the Dovetail® Omni-C® Kit (Dovetail Genomics, Cantata Bio) was used on the HMW DNA extracted from the liver, following the manufacturer’s protocol. After reversal crosslinking, the DNA was purified and biotinylated chimeric molecules isolated using streptavidin beads before PCR enrichment with 12 PCR cycles using KAPA HiFi HotStart Ready Mix (Roche). The short-insert paired-end library for whole genome sequencing was prepared using the PCR-free protocol and the KAPA HyperPrep kit (Roche). After end-repair and adenylation, Illumina™ platform-compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies) were ligated. The sequencing library was quality controlled on a 2100 Bioanalyzer using the DNA 7500 assay (Agilent), quantified with KAPA Library Quantification Kit (Roche), and sequenced on four lanes of a NovaSeq™ 6000 (Illumina) with a read length of 2×151 bp.
Total RNA from individual tissues (tail, gonad, heart, gill, skin, spleen, liver, kidney, and muscle) was used to prepare RNA-Seq tissue-specific libraries with a KAPA Stranded mRNA-Seq kit (Roche) following the manufacturer’s protocol. The transcriptomes were sequenced on 4 lanes of a NovaSeq 6000 (Illumina) with a read length of 2×151 bp.
Data generated with the PromethION and NovaSeq 6000 were assembled with the Centro Nacional de Análisis Genómico (CNAG) Snakemake pipeline v2.0 (https://github.com/cnag-aat/assembly_pipeline) to obtain an optimal base assembly for further Omni-C scaffolding. The list of programs, parameters and versions used to assemble and quality check the genome are presented in Table 1. In brief, Illumina reads were processed with Cutadapt (Martin, 2011), while ONT reads were filtered with FiltLong (https://github.com/rrwick/Filtlong). Filtered ONT reads were assembled with both Flye (Kolmogorov et al., 2019) and NextDenovo (Hu et al., 2024). GenomeScope2 (Ranallo-Benavidez et al., 2020; Vurture et al., 2017) was used to estimate genome size with the 20-mers present in the pre-processed Illumina reads. The NextDenovo (Hu et al., 2024) assembly was polished with both ONT and Illumina paired-end reads using Hypo (Kundu et al., 2019) and then the polished assembly was collapsed with purge_dups (Guan et al., 2020) to remove haplotypic duplications.
Dark grey: genome assembly. Light grey: genome annotation. Dark blue: assembly checks. Light blue: genome curation.
* Program ran within the Centro Nacional de Análisis Genómico (CNAG) snakemake pipeline v2.0 (https://github.com/cnag-aat/assembly_pipeline).
For further proximity ligation-based scaffolding, a total of 206.96 million Omni-C read pairs were mapped to the assembled genome using the Dovetail Genomics recommended protocol (https://omni-c.readthedocs.io/en/latest/fastq_to_bam.html). After excluding PCR duplicates, 106.91 million valid Omni-C read pairs were used to scaffold the assembly with YaHS (Zhou et al., 2023) using the default initial contig error correction step.
To guide manual curation of the assembly, the ONT read coverage was computed for all positions in the assembly using minimap2 (Li, 2018), SAMtools (Danecek et al., 2021; Li et al., 2009), and BEDtools (Quinlan and Hall, 2010), as well as the location of gaps with fasta-stats.py (https://github.com/cnag-aat/scripts/blob/main/fasta-stats.py) and telomeres with telomeric-identifier (Brown et al., 2023). These extensions were added to the contact map using PretextGraph (https://github.com/sanger-tol/PretextGraph). Manual curation was performed according to the rapid curation protocol from The Sanger Institute (https://gitlab.com/wtsi-grit/rapid-curation ) using PretextView (https://github.com/sanger-tol/PretextView). The genome was assessed for completeness with BUSCO using the odb10 Actinopterygii database (Manni et al., 2021), with Merqury (Rhie et al., 2020) for consensus accuracy (QV) and k-mer statistics, for contiguity statistics with fasta-stats.py (https://github.com/cnag-aat/scripts/blob/main/fasta-stats.py), and for contamination with BlobToolKit (with NCBI nt database, August 2023 update) (Challis et al., 2020) and BUSCO using the odb10 databases for Fungi and Bacteria (Manni et al., 2021). For comparison with the genome assembled in this study, the genome previously described by Li and colleagues (Li et al., 2023) and available in the European Nucleotide Archive (accession ID: PRJEB28248) was also assessed for completeness with BUSCO’s odb10 Actinopterygii database (Manni et al., 2021).
The hamour genome assembly annotation was obtained by combining transcript alignments, protein alignments and ab initio gene predictions. The list of programs, parameters, and versions used for genome annotation is provided in Table 1. In brief, repeats present in the genome assembly were annotated with RepeatMasker (Smith et al., 2007; http://www.repeatmasker.org) using the custom repeat library available for Danio rerio and a new repeat library specific for this study made with RepeatModeler (https://github.com/Dfam-consortium/RepeatModeler). After excluding repeats that were part of repetitive protein families from the resulting library, RepeatMasker (Smith et al., 2007; http://www.repeatmasker.org) was run again with this new library performing a BLAST (Altschul et al., 1990) search against UniProt (May 2023, https://www.uniprot.org/) to annotate the specific repeats. RNA-seq reads were aligned to the previously assembled genome using STAR (Dobin et al., 2013). Transcript models were subsequently generated using StringTie (Pertea et al., 2015) and merged using TACO (Niknafs et al., 2017). High-quality junctions to be used during the annotation process were obtained by running ESPRESSO (Gao et al., 2024) after mapping with STAR (Dobin et al., 2013). Finally, assembled spliced alignments were produced with PASA (Haas et al., 2008). TransDecoder (https://github.com/TransDecoder/TransDecoder) was run on the spliced alignments in PASA (Haas et al., 2008) to detect coding regions in the transcripts. The complete proteomes of Gymnodraco acuticeps, Sander lucioperca, Cottoperca gobio, and Perca fluviatilis were downloaded from UniProt (May 2023, https://www.uniprot.org/) and aligned to the genome using miniprot (Li, 2023). Ab initio gene predictions were performed on the repeat-masked assembly with GeneID (Alioto et al., 2018) and Augustus (with human parameters) (Stanke et al., 2006), and Genemark-ET in self-trained mode (Lomsadze et al., 2014) with and without incorporating evidence from the RNA-seq data. Finally, all the data were combined into consensus coding sequence models using EVidenceModeler (Haas et al., 2008). Additionally, untranslated regions (UTRs) and alternative splicing forms were annotated via two rounds of PASA (Haas et al., 2008) annotation updates. Functional annotation was performed on the annotated proteins with PANNZER’s online server (Törönen and Holm, 2022; http://ekhidna2.biocenter.helsinki.fi/sanspanz/).
ONT whole genome sequencing produced 137.75 Gb of data (coverage=128.62x) and Illumina produced 67.73 Gb of 2x151 bp pair-end reads (coverage=63.24x). Genome size (genome haploid length) estimated with GenomeScope2 ranged from 1,088,845,762 to 1,089,817,901 bp ( Table 2). For proximity ligation-based scaffolding, a total of 206.96 million Omni-C read pairs were mapped to the intermediate assemblies generated with NextDenovo (Hu et al., 2024), resulting in a final assembly with scaffold N50 of 45.64 Mb, N90 of 39.86 Mb and accounting for 1.07 Gb ( Table 3, Figure 1), consistent with the GenomeScope2 (Ranallo-Benavidez et al., 2020; Vurture et al., 2017) estimation. The assembled genome consists of 24 superscaffolds (making up 99.9% of the assembly) in accordance with the previously reported diploid karyotype (2n=48) for this species (Wang et al., 2010). It had a consensus accuracy of QV=47 and single-copy BUSCO gene completeness of 98.9% (BUSCO odb10 Actinopterygii) ( Table 3). No evidence of contamination was detected. Together, these statistics indicate that we have assembled a high quality, chromosome-level genome for the hamour.
Genome size estimated by GenomeScope2 (Ranallo-Benavidez et al., 2020; Vurture et al., 2017) on the pre-processed Illumina reads. bp: base pairs.
Genome completeness was assessed with BUSCO (Manni et al., 2021) using the Actinopterygii odb10 database updated on the 08/01/2024. Number of BUSCO groups searched for in the Actinopterygii database was 3,640. bp: base pairs; Mb: megabases.
| Attribute | This study | Li et al., 2023 |
|---|---|---|
| Genome assembly total length (bp) | 1,071,864,792 | 1,023,559,032 |
| Scaffolds number | 33 | 1450 |
| Scaffold N50 (Mb) | 45 | 2 |
| Contigs number | 140 | 159 |
| Contig N50 (Mb) | 18 | 2 |
| Completeness | 99.3% | 99.0% |
| Single-copy | 98.9% | 98.5% |
| Duplicated | 0.4% | 0.5% |
| Fragmented | 0.6% | 0.7% |
| Missing | 0.1% | 0.3% |
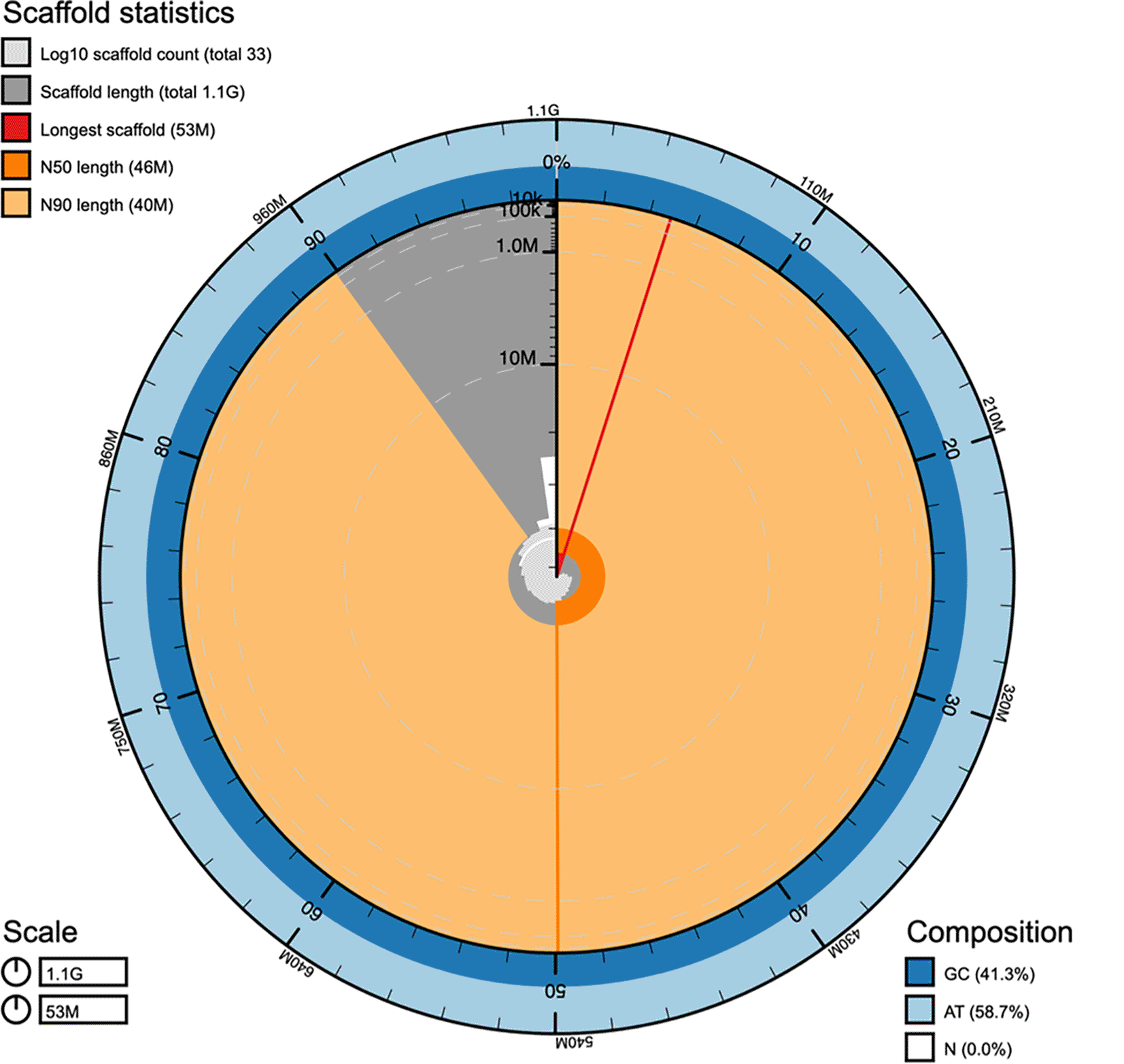
The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 1,071,864,792 bp assembly. The distribution of scaffold lengths is shown in dark grey with the plot radius scaled to the longest scaffold present in the assembly (52,562,209 bp, in red). Orange and pale-orange arcs show the N50 and N90 scaffold lengths (45,643,039 and 39,861,643 bp respectively). The light grey spiral shows the cumulative scaffold count on a log scale with white scale lines showing successive orders of magnitude. The blue and light-blue area around the outside of the plot shows the distribution of bases GC, AT, and N % in the same bins as the inner plot. bp: base pair; GC: guanine-cytosine; AT: adenine-thymine; N: nucleobase.
A comparison of the chromosome-level genome assembly produced in this study and a previously published genome (Li et al., 2023) is presented in Table 3. Genome size was consistent in both studies (1.07 and 1.02 GB in the present and previous study, respectively). Our assembly had a lower number of scaffolds with a greater scaffold length, providing an improvement on the previously published genome and contributing to the advancement of research for this species.
The genome annotation identified 28,384 protein-coding genes, producing 39,296 transcripts (1.38 transcripts per gene), which improved on the number of annotated protein coding genes reported in Li et al. (2023) (26,931). The annotated transcripts contained 11.05 exons on average, with 91% of them being multi-exonic ( Table 4).
The hamour genome assembly annotation was achieved by combining transcript alignments, protein alignments and ab initio gene predictions. The genome was annotated with 28,384 protein-coding genes, producing 39,296 transcripts (1.38 transcripts per gene). bp: base pairs; Mb: megabases.
Here we report on the sequencing and assembly of a hamour individual from the Arabian Gulf using a combination of Nanopore and Illumina sequencing technologies. We produced a chromosome-level assembly for this species and have improved on its annotation compared to a previously released genome. The genome sequence, raw data, and annotation are released openly for reuse. All raw sequence data, the assembly, and annotations have been deposited in INSDC databases, with accession identifiers reported in Table 5. These data will facilitate further studies on the biology of this species and on its management in the wild and aquaculture settings.
| Project accession data | |
| Assembly identifier | QU_Ecoi |
| Species | Epinephelus coioides |
| Specimen | QU-Ecoi-1 |
| NCBI Taxonomy ID | 94232 |
| BioProject | PRJNA1128520 |
| BioSample ID | SAMN42050860, SAMN43492902-SAMN43492913 |
| Isolate information | QU-Ecoi-1 |
| Raw data accessions | |
| Oxford Nanopore PromethION | SRR30574011 |
| Omni-C Illumina | SRR30574012 |
| Illumina short-read | SRR30574003 |
| Illumina RNASeq | SRR30574004-SRR30574010; SRR30574013-SRR30574014 |
| Genome assembly | |
| Assembly accession | GCA_051314025.1 |
Due the nature of the research project, with no experimental work on live animals (working only with tissues collected from dead animals), an exemption certificate from our institutional animal care and use committee (IACUC) was obtained for the use of wild fish caught by independent fishermen and bought by the author immediately after capture. The hamour used in this study was euthanised via immersion in MS-222 and destruction of the brain immediately after being caught. This study followed all relevant ethical guidelines and protocols approved by the institutional animal care and use committee (IACUC).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)