Keywords
PKHD1, Autosomal Recessive Polycystic Kidney Disease, Dhofar, Consanguinity, Genetic heterogeneity, Oman
This article is included in the Genomics and Genetics gateway.
PKHD1, Autosomal Recessive Polycystic Kidney Disease, Dhofar, Consanguinity, Genetic heterogeneity, Oman
Updated ethical approval reference number
To read any peer review reports and author responses for this article, follow the "read" links in the Open Peer Review table.
Autosomal recessive polycystic kidney disease (ARPKD) is one of the most common recessively inherited cystic kidney disorders, with an estimated incidence ranging from 1 in 6,000 to 1 in 55,000 live births (Zerres, Hansmann et al. 1988, Guay-Woodford, Bissler et al. 2014, Bergmann, Guay-Woodford et al. 2018). ARPKD most commonly presents in the neonatal and infantile period. It is characterized by progressive renal cystic dysplasia, which leads to enlarged, echogenic kidneys and varying degrees of renal impairment, as well as congenital hepatic fibrosis, which may progress to portal hypertension and its complications. Pulmonary hypoplasia due to oligohydramnios is a frequent and often life-threatening feature in perinatal presentations. While ARPKD is classically thought of as a disease of infancy and childhood, there is increasing recognition of later-onset and even adult presentations, in which renal and hepatic manifestations may be milder, slowly progressive, or discovered incidentally. The clinical spectrum therefore ranges from perinatal lethality to survival into adulthood with chronic kidney disease, portal hypertension, or combined hepatorenal involvement. While there is no definitive cure, current treatments focus on managing symptoms and delaying disease progression (Guay-Woodford, Bissler et al. 2014).
Disease-causing variants in PKHD1, which encodes the fibrocystin/polyductin protein, impair the development of the kidney and bile ducts. Defects in fibrocystin lead to kidney cyst formation and malformation of bile ducts in the liver, resulting in polycystic kidneys and congenital hepatic fibrosis. Truncating mutations in PKHD1 often result in severe neonatal outcomes, reflecting a genotype-phenotype correlation (Burgmaier, Brinker et al. 2021).
Although ARPKD has been widely studied in various populations, the genetic diversity of this disorder in Oman remains inadequately understood. Prior studies have identified several disease-causing variants in the PKHD1 gene within the Omani population, but these studies did not focus on the Dhofar region (Al Alawi, Molinari et al. 2020). The Dhofar region’s geographic isolation ( Figure 1), bordered by mountains and deserts, has fostered a unique demographic marked by consanguinity and minimal gene flow (Rajab, Hamza et al. 2015).
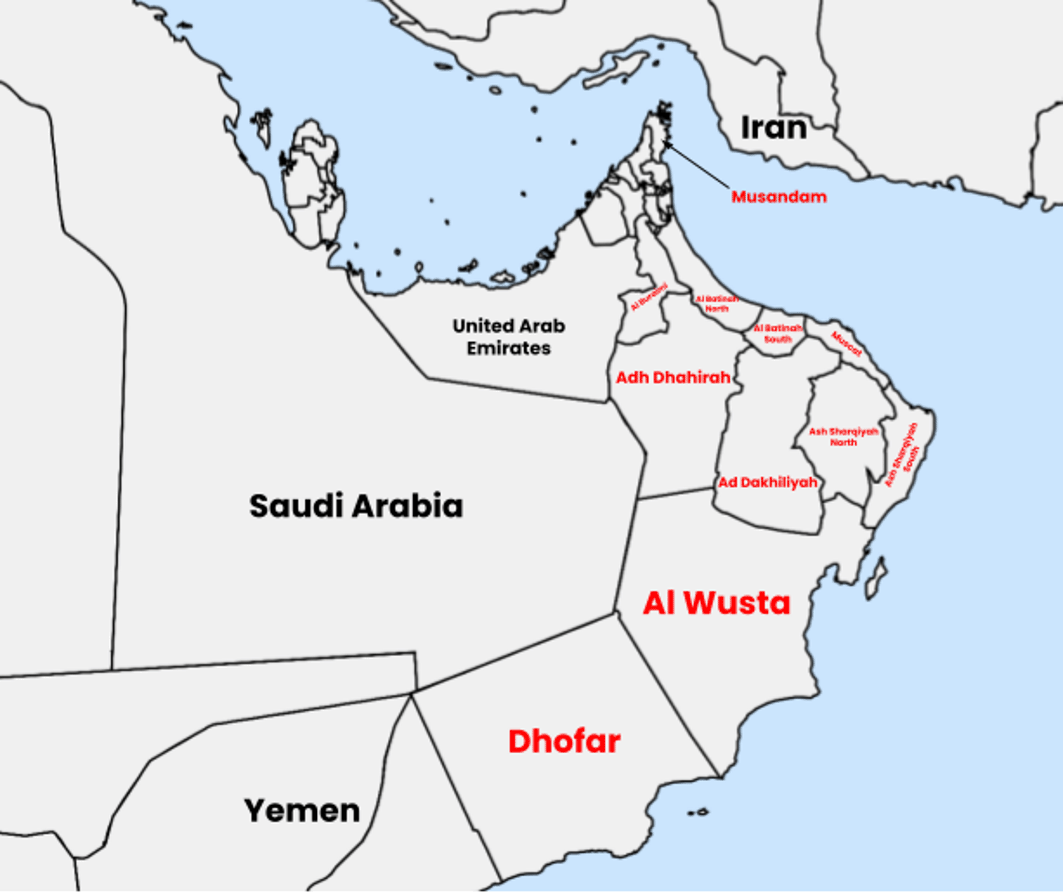
Map (drawn using https://www.mapchart.net/ and licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.) showing Dhofar region (Dhofar Governorate) in the south of Oman. It is the largest of 11 governorates in Oman in terms of area with a population of around 500,000 people.
Here we present a case series to investigate disease-causing PKHD1 variants identified in neonates from six families in the Dhofar region. The study aims to expand the genetic landscape of ARPKD in Oman and contribute to the broader understanding of this rare genetic disorder.
This case series includes six families from the Dhofar region of Oman who were referred to the National Genetic Centre at the Royal Hospital, Oman, following antenatal diagnoses of ARPKD between January 2023 and January 2025. All families had neonates with multiple congenital anomalies, including bilateral renal abnormalities, pulmonary hypoplasia, and oligohydramnios. This study was ethically approved by the Royal Hospital Scientific Research Ethical Committee, Ministry of Health, Oman (SRC/25647). For genetic studies of patients, written informed consent was provided by the parents of all affected neonates.
The six families included in this study were referred to the National Genetic Centre at Royal Hospital, Oman, based on antenatal findings of congenital anomalies consistent with ARPKD. The clinical features included: oligohydramnios, bilateral kidney enlargement, pulmonary hypoplasia, and multiple congenital anomalies (e.g., dysmorphic features such as low-set ears, box-shaped head, and sandal gap deformity). The clinical presentation, including prenatal ultrasound findings, birth details, and postnatal progress, was recorded. Neonatal deaths occurred within 48 hours postpartum in all cases, which provided an opportunity for genetic analysis from post-mortem DNA samples. Additionally, clinical investigations included antenatal ultrasound scanning where the prenatal diagnosis of ARPKD was based on ultrasound findings showing enlarged and echogenic kidneys, absent bladder, and oligohydramnios. Postnatal clinical observations were performed on arrival at the Special Care Baby Unit (SCBU), where the neonates were monitored for respiratory distress (grunting, cyanosis, and decreased oxygen saturation), and vital signs were recorded. The clinical progress for each family such as gestational age at delivery, birth weight and vital signs (e.g., heart rate, blood pressure, oxygen saturation, and temperature) was documented.
DNA samples for genetic analysis were extracted from cord blood/post-mortem blood specimens and archived at the National Genetic Centre for subsequent analysis. Clinical information was gathered from medical records, including antenatal ultrasound findings, birth outcomes, and postnatal management. Genetic testing was performed using both Sanger sequencing and next-generation sequencing (NGS) to identify mutations in the PKHD1 gene ( Al Alawi, Al Salmi et al. 2019). For Sanger sequencing, target regions were amplified using AmpliTaq Gold 360 Master Mix kit (Applied Biosystems) using gene specific oligonucleotide primers. For NGS, a multiplex PCR approach amplified 4898 amplicons from 60 target genes (including GANAB, HNF1B, NOTCH2, PKD1, and PKD2, among others) using a custom QIAseq panel (Qiagen). Subsequent NGS was performed on an Illumina MiSeq system. The resulting data (vcf files) was processed through Variant Studio software to allow nucleotide variant and copy number variant calling. All variant nomenclature followed the standards set by the Human Genome Variation Society guidelines and confirmed via Sanger sequencing. PKHD1 frameshift variants leading to predicted downstream nonsense alleles were modelled using MutationTaster2025 (https://www.genecascade.org/MutationTaster2025/) and sequences are shown in Extended Data.
The wild-type structure of fibrocystin was predicted with AlphaFold3 (Abramson, Adler et al. 2024) where the overall structure was of high confidence (pLDDT = 73.59). To model the frameshift and duplication variants, we predicted their complete structure and superimposed it on the wild-type. PyMOL was used for visualization (Schrödinger and DeLano 2020).
We analyzed six families with neonates affected by ARPKD. The clinical presentation of the affected neonates included signs of oligohydramnios, bilateral enlarged kidneys, pulmonary hypoplasia, and other dysmorphic features. All neonates died within 48 hours postpartum, with no survival beyond seven days. Table 1 illustrates the clinical presentation and outcomes of the affected neonates. The clinical progress for each family such as gestational age at delivery, birth weight and vital signs (e.g., heart rate, blood pressure, oxygen saturation, and temperature) was documented. The gestational age at delivery in our families ranged between 32 and 36 weeks, while the birth weight ranged from 2.3 kg to 3.4 kg. Table 2 summarizes these critical postnatal data.
Each of the six families described had a molecular genetic diagnosis of ARPKD secondary to biallelic PKHD1 disease causing variants ( Figure 2) and are summarized below.
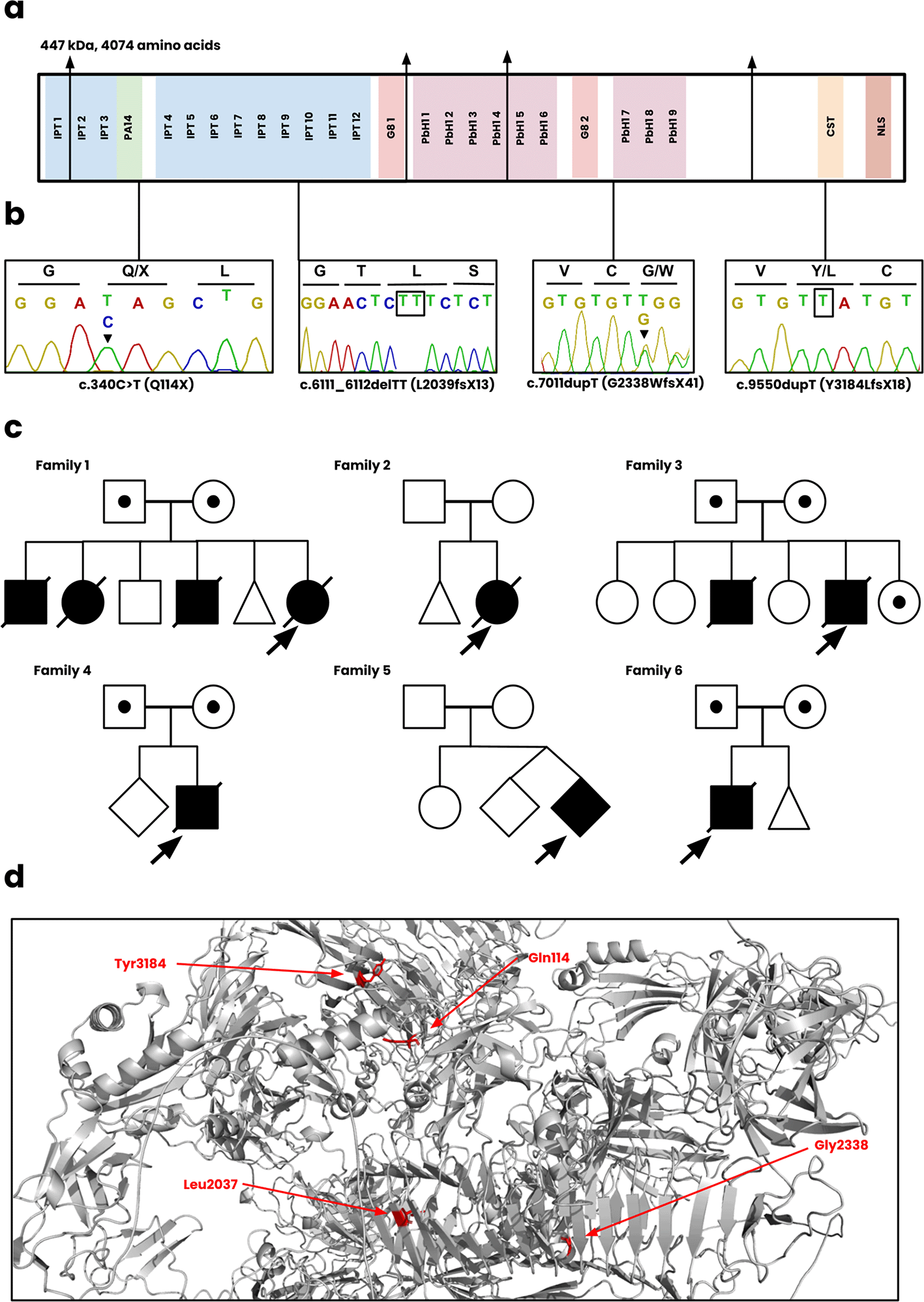
(a) The functional regions of fibrocystin, including the IPT, PA14, G8, PbH1, CST, and NLS domains. (b) Sanger sequencing of the c.340C>T, c.6111_6112delTT, c.7011dupT, c.9550dupT variants. The sequence for c.7011dupT of the patient is unavailable and instead displayed for the heterozygote parent. (c) The pedigree diagrams for all six Omani families. Squares, males; Circles, females; Shaded, affected. (d) Position of novel mutations on the wild-type structure of fibrocystin.
Family 1 included a female infant born at 34 weeks of gestation to consanguineous parents presented antenatally with bilateral enlarged kidneys and pulmonary hypoplasia. The infant died within 48 hours. Genetic testing identified a homozygous frameshift mutation, PKHD1 c.7011dupT; p.(G2338WfsX41), classified as likely pathogenic. There was a previous history of 3 neonatal deaths with similar clinical presentations.
Family 2 included a female infant, born at 33 weeks of gestation via breech delivery, had bilateral enlarged kidneys and pulmonary hypoplasia and died within 48 hours. Genetic analysis confirmed a homozygous frameshift mutation, PKHD1 c.7011dupT; p.(G2338WfsX41). There was a previous history of an early miscarriage.
Family 3 included a male infant, born at 35 weeks of gestation, was diagnosed antenatally with polycystic kidneys and suspected Potter syndrome. The neonate died within 7 hours due to spontaneous pneumothorax. Genetic analysis confirmed a frameshift mutation (not present in gnomAD, ClinVar, Leiden Open Variation Database) PKHD1: c.6111_6112delTT; p.(L2039fsX13).
Family 4 included a male infant born at 32 weeks of gestation to consanguineous parents presented with bilateral enlarged kidneys and other dysmorphic features. Genetic analysis revealed a novel homozygous deletion spanning exons 58-60 of the PKHD1 gene, classified as likely pathogenic.
Family 5 included a second twin from a consanguineous couple, born at 36 weeks, presented with oligohydramnios and bilateral kidney enlargement. The neonate died within 22 hours postpartum. A novel frameshift variant, PKHD1 c.9550dupT; p.(Y3184LfsX18), was identified.
Family 6: A male infant was born at 32 weeks of gestation to consanguineous parents presented with oligohydramnios and bilateral kidney enlargement. A homozygous nonsense variant, PKHD1 c.340C>T; p.(Q114*) was identified.
In Figure 3, we have displayed the predicted protein structures for the five novel PKHD1 variants superimposed onto the wild-type fibrocystin structure. The c.340C>T results in a nonsense Q114X variant that encodes for the complete IPT 1 domain ( Figure 3a) but causes a loss of most of its wild-type structure ( Figure 3b). The c.6111_6112delTT is predicted to cause a L2039fsX13 frameshift, impacting the G8 1 domain ( Figure 3c). The wild-type G8 1 domain is composed of 10 β-strands and one α-helix (He, Liu et al. 2006). In our structural model ( Figure 3d), we observed these structural characteristics, but an additional β-strand is formed resulting from the frameshift. The c.7011dupT mutation is predicted to result in a G2338WfsX41 frameshift, where the PbH1 domain is affected ( Figure 3e). The wild-type PbH1 domain is composed of 3 β-strands stacked into six repeats (Mayans, Scott et al. 1997). In the mutant structure, PbH1 1 to PbH1 4 is normal, but the frameshift results in a loss of the remaining two repeats and instead a structure composed of a loop and α-helix is formed ( Figure 3f). Lastly, the c.9550dupT (Y3184LfsX18) is located on an unknown region of fibrocystin ( Figure 3g), where the frameshift does not form secondary structure, and the C-terminal region is lost ( Figure 3h).
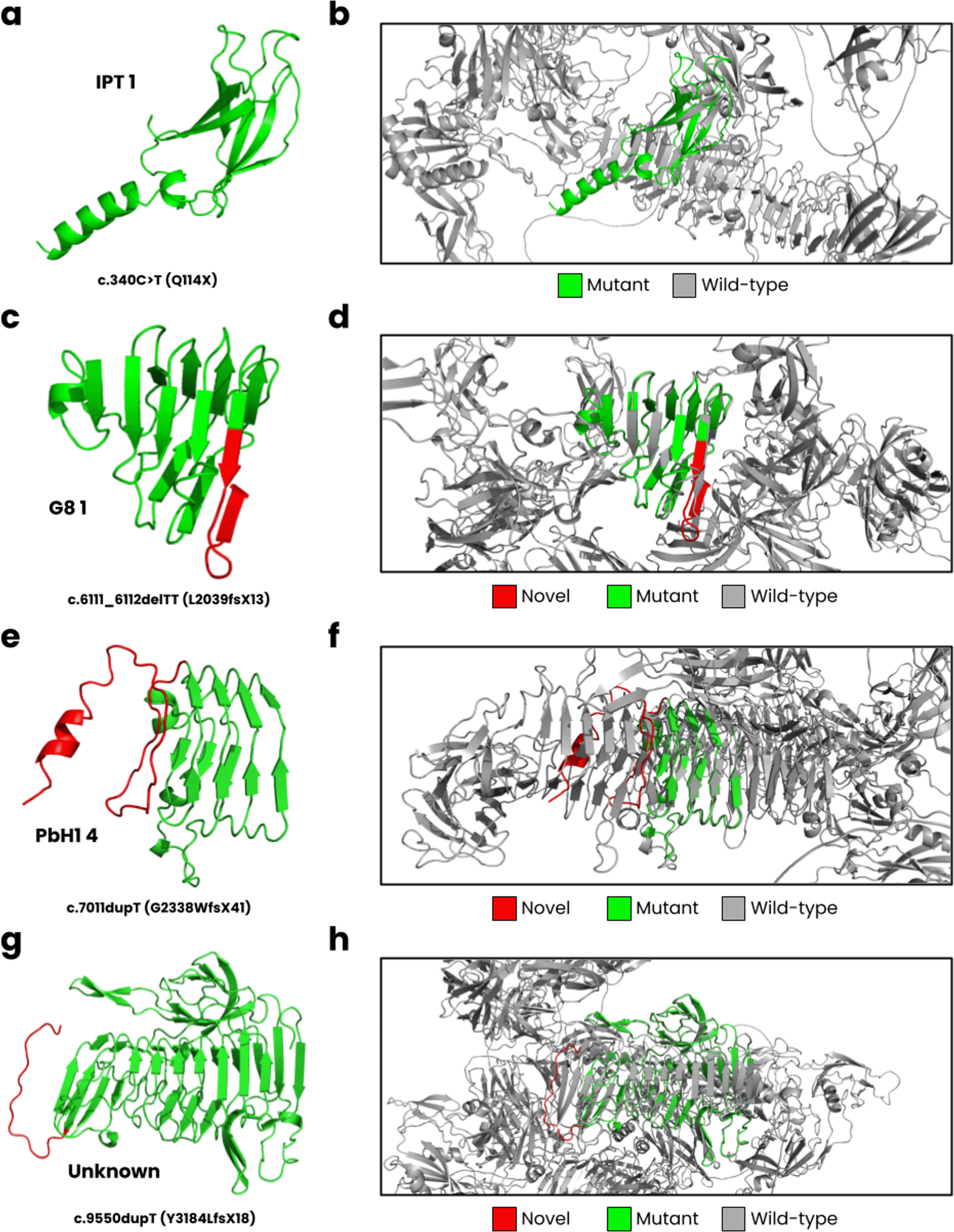
(a) The c.340C>T mutation is predicted to cause a Q114X nonsense variant, (b) where the mutant structure only partially encodes for the IPT one domain compared to the wild-type structure. (c) The c.6111_6112delTT leads to L2039fsX13, impacting the G8 1 domain, and (d) encoding nearly half of the wild-type structure. (e) The c.7011dupT cause a G2338WfsX41 frameshift, (f ) where PbH1 is partially lost. (g) Lastly, the c.9550dupT leads to Y3184LfsX18, encoding an unknown domain of PKHD1 (h) close to the C-terminal region.
The universal presence of pulmonary hypoplasia in these neonates aligns with ARPKD’s hallmark features and is often associated with Potter syndrome due to oligohydramnios (Guay-Woodford, Bissler et al. 2014). The severity of oligohydramnios and lung underdevelopment could explain the early mortality observed in these cases. Despite genetic heterogeneity, the clinical presentations were remarkably consistent, with common features such as bilateral kidney enlargement and dysmorphic features. This could point to a shared pathophysiological mechanism linked to the PKHD1 mutations in this population.
This case series highlights the identification of five distinct mutations in the PKHD1 gene in neonates from the Dhofar region of Oman. The presence of a recurrent variant (c.7011dupT), a nonsense variant and three other novel variants (c.6111_6112delTT, c.9550dupT, and a large deletion) suggests significant genetic heterogeneity within the region. Notably, these alleles were not previously reported in studies of ARPKD in other parts of Oman (Al Alawi, Molinari et al. 2020), indicating that the Dhofar population may harbor unique genetic variants. While previous studies of ARPKD in Oman have focused on more populous regions, this study uncovers significant genetic diversity within the Dhofar population. The identification of novel mutations, including c.6111_6112delTT and c.9550dupT, points to the complex genetic architecture of ARPKD in Oman, which may be influenced by regional variations in genetic isolation and migration patterns.
The Dhofar region, the southernmost governate of Oman, is geographically isolated by mountains and deserts, which likely restricts gene flow from neighboring populations. This isolation can contribute to the accumulation of distinct genetic mutations, as observed in the c.7011dupT allele in two families, who are likely to be related. This was also true for a CFTR allele reported in patients with cystic fibrosis that was also specific to this distinct geographical region (Al Oraimi, Al Shidhani et al. 2022). The high rate of consanguinity in Dhofar (up to 68%, (Al Oraimi, Al Shidhani et al. 2022)) likely enhances the likelihood of genetic drift and the founder effect, potentially explaining the presence of this mutation in multiple families.
This study identifies five distinct PKHD1 mutations in neonates from the Dhofar region of Oman, contributing to the genetic understanding of ARPKD. Our findings underscore the need for regional studies to map Oman’s genetic diversity. Further research, including larger studies involving other regions of Oman, is necessary to better understand the molecular basis of ARPKD in this population and to inform genetic counseling and management strategies for affected families.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)