Keywords
Population frequency, Structural variants, Mosaicism, Cancer, LLM, Metagenome, tandem repeats, haplotype structure, ancestral recombination graphs
This article is included in the Genomics and Genetics gateway.
This article is included in the Bioinformatics gateway.
This article is included in the Hackathons collection.
Population frequency, Structural variants, Mosaicism, Cancer, LLM, Metagenome, tandem repeats, haplotype structure, ancestral recombination graphs
Baylor College of Medicine hosted the sixth annual structural variant and pangenomics Hackathon on August 28th-30th, 2024. We reported the results of some of the previous hackathons as articles (Deb et al. 2024; Walker et al. 2022; Mc Cartney et al. 2021).
Tandem repeats (TRs) are DNA sequences consisting of two or more bases repeated multiple times in a head-to-tail pattern along a chromosome (Levinson 2019). Typically found in non-coding regions, TRs play significant roles in genetic variation and are implicated in various diseases (Depienne and Mandel 2021). They also serve as powerful tools in DNA fingerprinting for forensic analysis (Butler 2006).
TR subtypes are classified based on the length of the repeated motif—short tandem repeats (STRs) range from 2 to 6 base pairs (Butler 2006), while variable number of tandem repeats (VNTRs) span 7 to 100 base pairs (Bakhtiari et al. 2021). Additionally, TRs can be categorized by their genomic context or function, such as alpha satellite repeats in centromeres (McNulty and Sullivan 2018; A. English et al. 2023) or rDNA repeats (Kobayashi 2014). Despite their importance, tandem repeats are challenging to analyze.
Our project aims to leverage the existing Tandem Repeat Database and Analysis Queries tool (tdb). This tool turns ‘REPL’ style VCFs from tandem repeat (TR) callers into a database. This database is in parquet format, compressed and well-structured and easily parsed as compared to VCFs. There are currently a handful of ‘standard’ queries and analysis notebooks which can provide useful summaries of tandem repeat results. For the Sixth Annual Structural Variant and Pangenomics Hackathon, we aimed to introduce some new and interesting queries.
Mosaic variants are genetic mutations that affect only a subset of an individual’s cells rather than all of them (Jiang et al. 2019). This mutation occurs after fertilization and during early development, resulting in a mosaic pattern in which some cells carry the genetic change while others do not. Mosaic variants can affect multiple tissues and produce a wide range of phenotypes, depending on when and where the mutation occurs during development (Biesecker and Spinner 2013).
Mosaic variations help explain the genetic risk of adult diseases. It’s vital to understand their normal, non-pathogenic incidence and mutation rates (Costantino, Nicodemus, and Chun 2021). To do this, we primarily use variant detection approaches across sequencing platforms. We customize some approaches for specific conditions, which complicates the evaluation of their accuracy and false positive rates. We created a modeling framework to mimic mosaic mutations at varied variant allele frequency (VAF) rates, including substitutions, indels, and structural variations. Our project builds on the group project from last year, ensuring that it is suitable for long-read sequence files. The identification of mosaic mutations is often based on the analysis of VAF, which reflects the proportion of sequencing reads that have a particular variant. Detecting variants with low VAF can be difficult as they may be masked by sequencing errors or only occur in a small number of cells. This becomes particularly important when studying complex diseases or conditions where a subtle mosaicism might influence disease onset or progression.
Antimicrobial resistance (AMR) is a growing global health concern, driven by the overuse and misuse of antibiotics (Sugden, Kelly, and Davies 2016). Detecting and monitoring the presence of AMR genes in various environments is crucial for understanding the spread of resistance and informing public health strategies. Matching metagenomes with AMR genes makes it easy to survey a wide range of samples at once, when compared with wet lab methods. This has been a popular field so multiple tools and databases were published. However, we still have some unexplored areas regarding databases.
Finding AMR genes usually involves two data components, a database of known AMR genes and the database which they search. The search is fulfilled by alignment tools, including DIAMOND (Buchfink, Xie, and Huson 2015), BLAST+, HMMER or minimap2 (Li 2018). For example, AMRFinder and ARGminer are two approaches with specific purposes. These tools usually accompany curated standard databases of AMR genes. For example, the Comprehensive Antibiotic Resistance Database (CARD) is a systematically maintained database that combines the Antibiotic Resistance Ontology (ARO) with well curated AMR gene sequences and mutations (Alcock et al. 2023). The database offers a methodical approach to categorize and comprehend resistances, using separate files for each model type, FASTA data, and ARO tags linked to GenBank accessions, and also offers cross-references for primary categories within the ARO, such as the AMR gene family, target drug class, and resistance mechanism.
Regarding genomic and metagenomic sequences, European Nucleotide Archive (ENA) Repository and National Center for Biotechnology Information (NCBI) GenBank were common modern choices. In history, the quantity of publicly available genomic and metagenomic sequences has grown exponentially since the beginning of centralized cloud-based hosts for genetic sequencing data (Lathe et al. 2008). The Los Alamos Sequence Database was first established as a repository for annotated biological sequences in 1979, and then relocated to the National Center for Biotechnology Information (NCBI) and renamed to GenBank in 1982 (“GenBank and WGS Statistics” 2024; “National Library of Medicine” 2024; Sayers et al. 2020). This database is now part of the International Nucleotide Sequence Database (INSDC), a collaboration between NCBI, the European Molecular Biology Laboratory (EMBL) and the DNA Databank of Japan (DDBJ), and as of August 2024, contains roughly 3.68 terabases of sequencing data (“GenBank and WGS Statistics” 2024). In 2009, INSDC additionally launched Sequence Read Archive (SRA) to host raw and unprocessed reads (Katz et al. 2022). As of December 2023, this massive dataset contains roughly 50 petabases of sequencing data from a range of eukaryotic and prokaryotic hosts, as well as environmental communities (Chikhi et al. 2024). Each set of sequences contains metadata subject to that of the uploader with information regarding the sequencing process (i.e. assay type, sequencing instrument, library layout) as well as the sampled environment (i.e. organism, sampling date, geographical location).
However, that most resourceful database, SRA, was difficult to search because of its size. In 2024, a nearly comprehensive solution, called Logan, was published on BioRxiv. The Logan database consisted of assembled contigs and unitigs, derived from a freeze of the SRA, reduced the size and redundancy of raw reads (384 terabytes vs. 50 petabases). Longan permits large-scale alignment-based search across all sequences on SRA across the Tree of Life efficiently for the first time. These constructed assemblies in conjunction with Amazon Web Services (AWS) structure permit large-scale alignment to set of query protein or nucleotide sequences, via DIAMOND (Buchfink, Reuter, and Drost 2021) or minimap2 (Li 2018) respectively, within a reasonable amount of time. Leveraging the vast quantity of unprocessed data on SRA rather than smaller annotated databases, gives potential to detect large-scale trends of gene flow around the globe across different organisms and environments.
In this research, we aligned the genes of CARD to the Logan database to identify and catalog AMR genes present in the dataset. Therefore, we could survey the prevalences, mechanisms, distributions and other important properties of antimicrobial resistance. This work will provide valuable insights into the distribution and prevalence of AMR genes across a vast range of environments and host organisms. This approach surpasses previous attempts to find AMR in SRA subsets by taking advantage of the sheer size of the Logan database, which contains all accumulated information from SRA to date, and by using contigs for alignment, which should help avoid issues with contaminations faced by raw reads.
Mobile genetic elements (e.g., transposons) are capable of relocating within a genome through cut-and-paste and copy-and-paste mechanisms. Their movement can influence gene expression, exert mutagenic effects, and drive genome evolution. In humans, they are implicated in the origin of diseases (Chénais 2022). Conversely, they hold potential for use in genetic editing, particularly in the treatment of genetic disorders, thereby underscoring the importance of their identification and annotation within the genome. In fungi, transposons confer metal resistance and contribute to genome evolution. However, the identification and annotation of mobile elements present considerable challenges due to their structural diversity, which complicates genomic mapping. Additionally, their capacity for horizontal transfer between species further complicates the determination of their function.
Starfish (Gluck-Thaler and Vogan 2024) is a recently developed modular toolkit for de novo giant mobile element discovery and annotation in fungal genomes. In an effort to support the use of starfish for other species, this project aimed to bolster accessibility and usability of starfish (v1.0.0).
Oxford Nanopore (ONT) sequencing is rapidly becoming a widely used sequencing technology in metagenomic studies due to its cost, long reads, and significantly improved error rate (Agustinho et al. 2024). However, there exists a wide heterogeneity in microbiome data due to variation in experimental designs making designing efficient computational software challenging. As long reading sequencing technology becomes popular in metagenomics, simulated datasets with known error rates can help evaluate existing and newer bioinformatic algorithms. There is a need for an easy to use metagenomic tool development to create standard truth ONT datasets in varying microbial environments that are reasonably realistic. We built MIMIC, a metagenome simulator that creates simulated ONT sequencing data by replicating the taxonomic abundances of real-world microbiome samples. In addition to providing simulated sequencing data, MIMIC also offers a simple-to-use evaluation framework for comparing the results of existing taxonomic classification methods to the known truth data, allowing for easy benchmarking across a host of different environments and error-profiles.
Haplotypes are defined as sets of genomic variants that are inherited together from a single parent. In theory, the human genome consists of multiple haplotype blocks shared among individuals from all populations, however, there are differences in allele frequency between any two populations (Shipilina et al. 2023). Haplotype phasing estimates the haplotype inheritance using genotype or sequencing data and aims to capture information about which genomic variation is associated with particular complex traits and common diseases, such as cancer (Garg 2023; Sakamoto, Sereewattanawoot, and Suzuki 2019) or diabetes (Sankareswaran et al. 2024; Luo et al. 2024). By estimating haplotypes, we can infer inter- and intra-population genealogical relationships, thus enhancing our understanding of the relatedness among individuals in the population, as well as the implications of a given mutation (or variation) on health.
In recent years, global initiatives have been undertaken to determine genomic variation that underlie phenotypic similarities across different populations, such as the International HapMap Project (“The International HapMap Project” 2003). Furthermore, the increasing number of biomedical databases, such as 1000Genomes (“A Global Reference for Human Genetic Variation” 2015), Genome in a Bottle (Zook et al. 2016) or UK Biobank (“UK Biobank” 2024) provide access to large collections of genomic data which can advance the efficiency and accuracy of methods for variant phasing and genealogical analyses. However, accurately estimating haplotypes and interpreting their implications in the disease mechanisms remain challenging due to the complexity of the data and high computational cost, therefore previous approaches for haplotype analysis would make broad assumptions and rough approximations, which could lead to inaccuracies. Conversely, new approaches for inferring the association between genomic variation and complex traits, alongside a large-scale computing infrastructure offer an amazing opportunity to efficiently and accurately derive genealogical relationships, ancestry, causality and risk factors for shared phenotypic traits (Browning and Browning 2023; Hofmeister et al. 2023; Leitwein et al. 2020).
During the hackathon, we aimed to design and develop a bioinformatic analysis pipeline for the computation of similarity matrices of intra- and interpopulation haplotype blocks, which would take into account both rare and common genomic variants. Here we present a proof-of-concept bioinformatic workflow to obtain haplotype blocks and to determine correlations between sets of genomic variants and genealogical relationships. We planned to use the existing methodologies for haplotype phasing, SHAPEIT5 (Hofmeister et al. 2023), and relatedness calculation, ARG-Needle (B. C. Zhang et al. 2023), to examine how sets of genomic variants are shared across populations. We used ARG-Needle (Zhang et al., 2023) to infer genealogical relationships between two haplotype blocks: a haplotype block that overlaps with the human leukocyte antigen HLA-A gene (chr6:29631001-30180001) and a random haplotype block (chr6:594001-655001). Then we planned to use these evolutionary relationships in the form of ancestral recombination graphs (ARGs), which offer a promising direction in evolutionary research (Griffiths and Marjoram 1997; Lewanski, Grundler, and Bradburd 2023) to estimate similarities between the haplotype blocks across populations.
Cancer is a highly heterogeneous microevolutionary state that arises from healthy cells by a series of point mutations and large DNA rearrangements. Sporadic mutagenesis gives rise to tumor subclones that have a distinct set of genomic alterations, which can promote tumor growth, metastasis or treatment resistance. In comparison to single nucleotide variants, characterisation of more complex events that contribute to intratumoral genetic heterogeneity was lacking up until recent efforts in deep whole genome sequencing of tumors and development of mutation callers. In this project we specifically focus on mosaic structural variants in cancer and have designed a tool for their functional annotation for identifying which genes and biological pathways are affected by these mosaic structural variants. Therefore, by comprehension and linking genes we can predict how a tumor might evolve over time, by extension this leading to mutation prediction which has aggressive tumor behavior or how cancer might respond to different treatments. While simple, this tool should become a stepping stone for further studies on the contribution of rare variants to emergence of treatment-resistant subclones and the recurrence of disease.
Structural variants (SVs) represent deviations from a reference genome sequence, typically spanning more than 50 base pairs (bps). These variations can have significant implications for understanding genetic diversity and the mechanisms underlying various phenotypes. Larger structural variants are present among human genomes. In particular, human chromosomes can have deletions of segments, duplications of segments, inverted segments, inserted segments, and/or translocated segments from other chromosomes ( Figure 1).
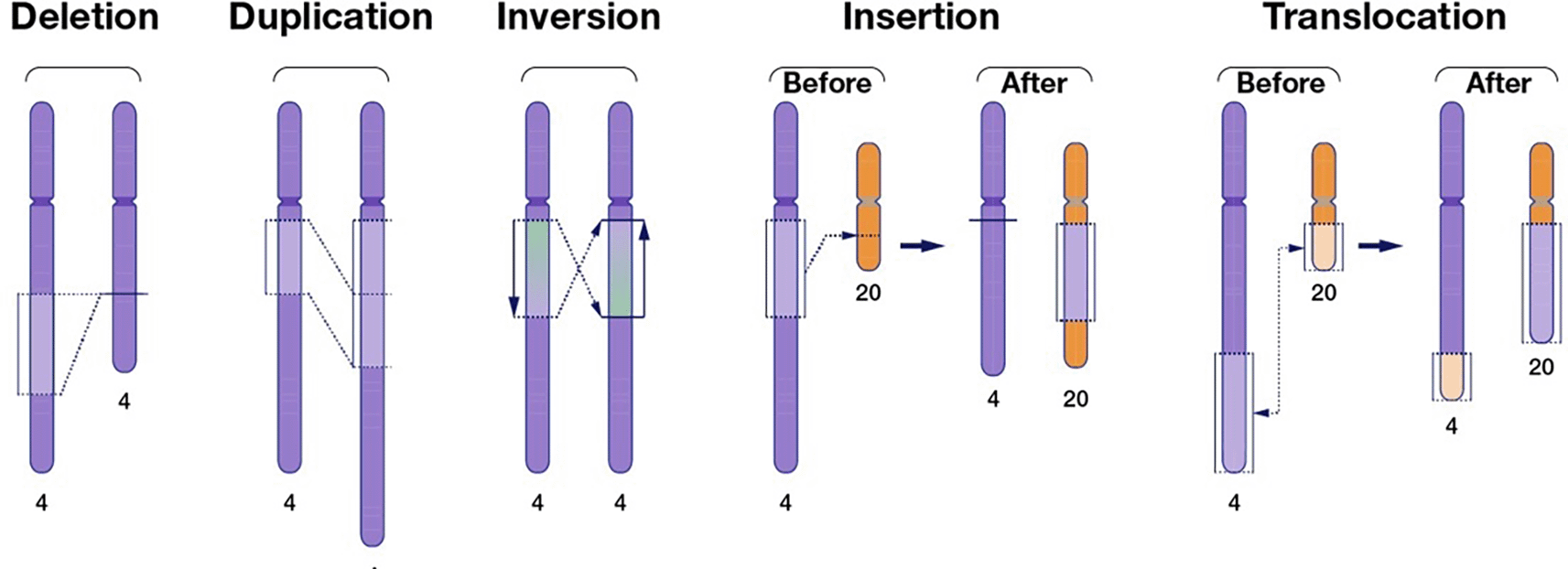
For example, the Charcot-Marie-Tooth disease type 1A (CMT1A) that results in nerve damage in extremities is caused by a duplication of the peripheral myelin protein 22 (PMP22) gene on human chromosome 17 (Lupski et al. 1991; Stavrou and Kleopa 2023). This condition is prevalent among at least 17 out of every 100,000 people worldwide as a result of the same SV on the PMP22 gene (Ma et al. 2023). By localizing the affected region, animal models in preclinical trials hope to completely reverse the condition through gene silencing.
This project aims to develop a robust pipeline for detecting and cataloging identical SVs across different samples and databases, ultimately linking them to specific phenotypes. The primary goal of this study is to identify and analyze SVs in novel and known genes, as well as established population SVs, to uncover new biological processes and associations. By cross-referencing SVs with phenotypic data, this pipeline seeks to establish a more comprehensive understanding of genotype-phenotype correlations.
Data
We used a tandem repeat database (TDB) containing 105 samples of diverse ancestries from the Human Pangenome Reference Consortium (HPRC) (Liao et al. 2023; Dolzhenko et al. 2024). The population distribution of 105 individuals from the TDB database include 52 African ancestry (AFR), 56 American ancestry (AMR), 32 East Asian ancestry (EAS), 48 South Asian ancestry (SAS), and 8 unknown ancestry (UNK). The data encompasses 937,122 tandem repeat (TR) loci spanning a total of 121,698,022 base pairs, which represents approximately 4% of the GRCh38 reference genome. Additionally we used the Adotto TR catalog (v0.3) (“Project Adotto Tandem-Repeat Regions and Annotations” 2024).
Queries
We had four queries for the completion of the hackathon project ( Figure 2).
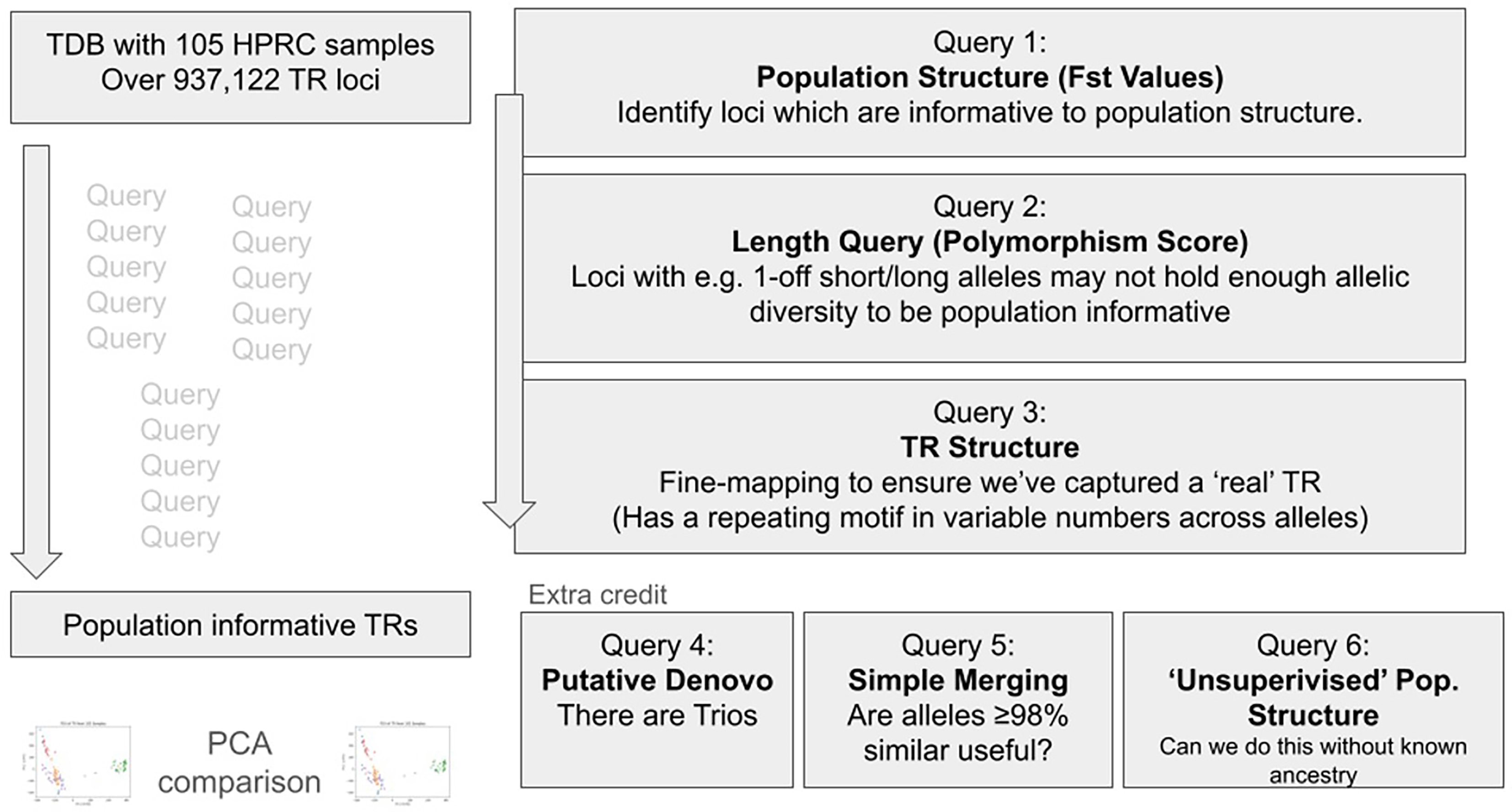
During the hackathon, four queries were completed: GTF annotation, population structure and PCA, outlier length, and TR structure.
First query was a GTF annotation. There is an established population structure notebook (https://github.com/ACEnglish/tdb/blob/develop/notebooks/PopulationStructure.ipynb) which will identify loci with >= 20 alleles and plot a clustermap of how similar samples’ alleles are. This comes with clustering in the HPRC example data which constructs the population structure. This query selects the loci which is greater or equal to 20 alleles sufficient and leverages the length of polymorphism queries to get an informative set of loci. This query also further includes samples with their clusterID which reveals more information for understanding population structure. Second query was to study population structure and PCA analysis. Though there is already an example notebook which will perform a PCA on a tdb. This query can be expanded to perform PCA on methylation data and relate population structures to its methylation data. Third query was about length outliers. We used this approach to find TR alleles which have an anomalous length and to explore length outliers. This query will help to incorporate other approaches to find length outliers. Finally, the fourth query was about the TR structure. Given the multiple TR alleles over a locus, we can annotate the TR motifs on each sequence and perform an MSA. We can then consolidate and create a ‘consensus’ structure of the repeats over the spans. This output should allow more detailed analysis of length outliers because we would no longer be just looking at the length of sequence over the locus but have motifs and copy numbers aligned across alleles. A light-weight notebook that leverages abpoa and tr-solve to build some of this information is already available. However, we want to replace tr-solve for annotating motifs. TRF is possible, but it will redundantly annotate spans which would make deconvolution of the repeat structure over multiple sequences difficult.
Implementation
We used tdb v0.2.0, which creates and analyzes genomic databases that have tandem repeat sequences. It is available through tdb github release.
Operation
We installed tdb by cloning its repository and installing it via Python. To process the data we created a tdb-compatible file from a VCF and queried allele counts, using the create and query commands. We merged tdb files using the merge command, which combines two databases with higher memory allocation. Additionally we added extra files using the merge --into option. For larger datasets containing more than ten tdb files, we used bigmerge command in order to effectively query and manage tandem repeat databases.
Our simulation framework models mosaic mutations at various variant allele frequency (VAF) rates, including substitutions, indels, and structural variants, using two tools: SpikeVar and TykeVar.
• SpikeVar automates the merging of two datasets at user-defined coverage or rates, verifies variant-calling mutations, and outputs a benchmarking-ready VCF file with accurate VAF annotations.
• TykeVar modifies reads within a single sample to simulate mosaic mutations while preserving haplotype structures. Altered read IDs are removed from the BAM file, aligned to the reference genome, and merged back. The final output consists of a modified BAM file and a VCF file with annotated mosaic variants.
As illustrated in Figure 3, the SpikeVar pipeline generates a BAM file containing mixed sequencing reads. These reads originate from two samples combined in user-defined ratios, simulating mosaic VAF. The resulting VCF file annotates confirmed mosaic variant locations, providing variant positions and supporting information. The TykeVarMerger refines these outputs by integrating modified reads into the dataset, resulting in a filtered BAM with the original read IDs and a VCF containing verified mosaic variant records.
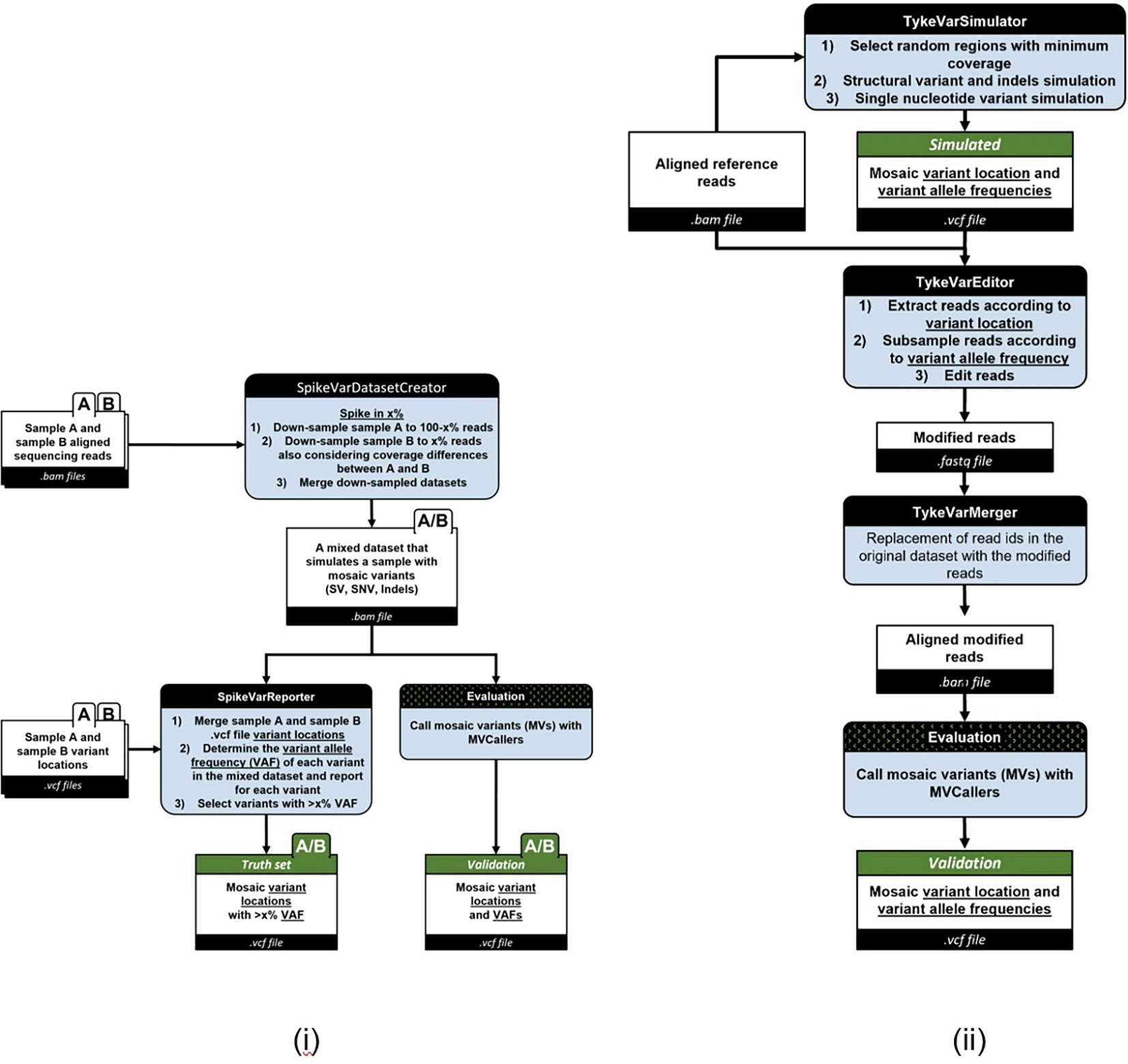
(A) The SpikeVar pipeline simulates mosaic variant allele frequency (VAF) by spiking mutations from one sample into another, creating a mixed dataset for variant callers. (B) TykeVar: A pipeline that inserts mosaic mutations into single-sample reads to create a modified dataset with original mosaic variations for accurate variant detection.
This integrated framework ensures reproducibility, scalability, and compatibility across sequencing datasets, facilitating robust and accurate benchmarking of mosaic variant detection methods (Deb et al., 2024).
Implementation
Our simulation framework integrates two primary tools, SpikeVar and TykeVar, to model mosaic mutations.
• SpikeVar automates the merging of datasets, verifies variant-calling mutations, and generates VCF files with accurate VAF annotations. It employs scripts such as 2b_regenotyping_main.sh, 2b_SNV.sh, 2b_SV.sh, 2b_vf_short.sh, 2b_vf_long.sh, 2b_vaf_filtering.sh, and 2b_vaf_merge.sh for distinguishing SNVs and SVs, processing sequencing data, and generating merged VCF outputs ( Figure 3(A)).
• TykeVar modifies reads in single-sample datasets, removing altered read IDs, aligning modified reads to the reference genome, and merging them back into the dataset. This results in BAM and VCF files with accurate truth sets for mosaic variants ( Figure 3(B)).
Both tools work together to create reliable datasets for benchmarking mosaic variant detection.
Operation
The following are the minimal system requirements and an overview of the workflow for running the SpikeVar and TykeVar pipelines:
System Requirements:
• Operating System: Linux (Ubuntu 20.04 or later recommended)
• Processor: Multi-core CPU (Intel Xeon or equivalent recommended)
• Memory: Minimum 64 GB RAM (128 GB recommended for larger datasets)
• Storage: At least 1 TB of free disk space
Software Dependencies:
Bash shell
Python (version ≥3.8)
SAMtools (version ≥1.10)
BCFtools (version ≥1.10)
BEDTools (version ≥2.30)
Variant callers (e.g., Mutect2, FreeBayes)
Workflow overview
SpikeVar Workflow:
1. Start with 2b_regenotyping_main.sh.
2. Process SNVs (2b_SNV.sh) and SVs (2b_SV.sh).
3. Use 2b_vf_short.sh or 2b_vf_long.sh for short or long-read processing.
4. Apply VAF filtering (2b_vaf_filtering.sh).
5. Generate the final “Merged Re-genotyped VCF” (2b_vaf_merge.sh).
TykeVar Workflow:
1. Remove altered read IDs from the original BAM file.
2. Align modified reads to the reference genome.
3. Merge modified reads into the filtered BAM.
4. Generate final BAM and VCF files with mosaic variant truth sets.
These workflows ensure reproducibility, scalability, and compatibility across sequencing datasets, facilitating accurate benchmarking of mosaic variant detection tools.
The prokaryotic subset of the Logan database was downloaded on 25 August 2024. Additionally, the CARD (version-3.3.0) database containing curated sequences of known AMR genes was obtained. Sequences from the CARD database were aligned to the Logan unitigs/contigs by minimap2 (Li 2018) with default parameters and the following arguments: `--sam-hit-only` and `-a`. We focused on high-confidence alignments that suggest the presence of AMR genes. Then, the results were filtered and curated using the NM tag in the SAM format, considering matches of at least 100 bases and identity of 80 bases. We benefited from the metadata of SRA accessions including location and date of samples.
We identified the number of alignment hits of AMR genes in the isolates over years from 2000 to 2024 in the United States. We visualize the results spatially using geopandas (v1.0.1) and mpl_toolkits from Matplotlib (v3.8). The workflow of the project is presented in Figure 4.
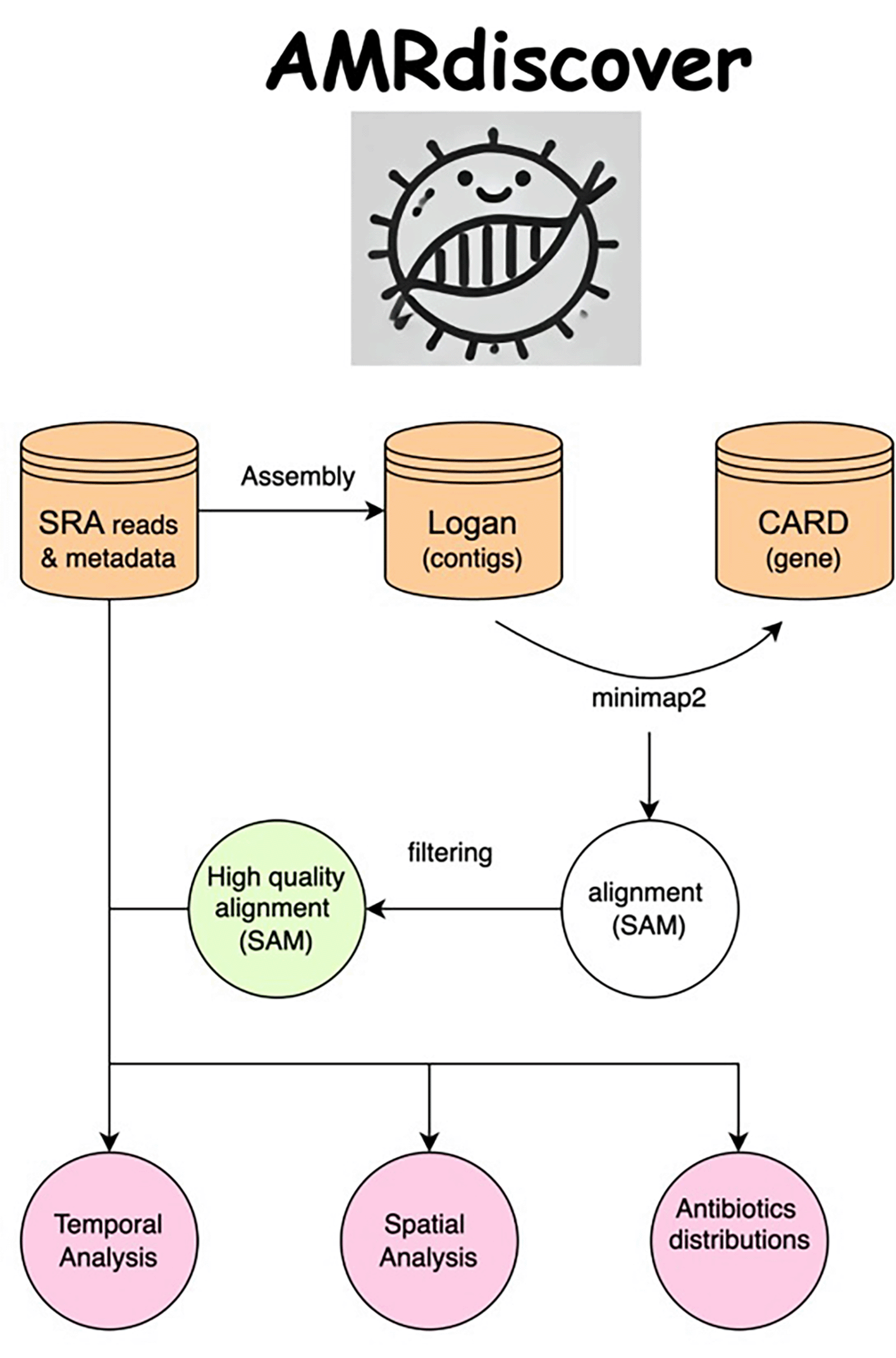
The pipeline consists of steps for analyzing the whole SRA database using the LOGAN contigs and CARD AMR genes. The output results are reported as the number of alignment hits for a country in a certain year.
Data
The Longan unitigs are available on AWS (https://registry.opendata.aws/pasteur-logan/). The antibiotic resistant genes were downloaded from CARD website ( https://card.mcmaster.ca/download/).
Implementation
Basically, our project was composed of three parts, alignment, filtering and analysis. The alignment was done with default minimap2. After alignment, the files were downloaded to local with AMRdiscover.sh and filtered by filter_parse_script.sh. The old_alignment_parsing.sh included the parallelization of filter_parse_script.sh. The analysis was diverse because each part was completed by different members. Geographic visualization was done with Python and the scripts were stored on our Github page, “AMRdiscover/scripts/sql_Athena”. Other scripts for visualization were done with Python3.8 or R4.2. For instance, species_gene_counts_plots.ipynb and plotting_mechanism.R.
Operation
Our analysis was performed on Linux Ubuntu 20.04. Alignment and its processes required minimap2 and samtools. Key tools on Linux were “awk” and GNU “parallel”. We used Python 3.8 and the following packages: pandas, matplotlib, geopandas, numpy, mpl_toolkits.axes_grid. R packages were tidyverse, RColorBrewer and khroma.
The current starship analysis (v1.0.0) requires executing seven individual bash scripts (https://github.com/egluckthaler/starfish). To simplify execution, starfishDiscovery provides a docker container.
Implementation
To use the Docker container, clone the starfish repository and from the same directory as the Dockerfile, run the following command: `docker build -t ${docker_username}/starfish --platform linux/amd64`. This will build a Docker container that includes all the software needed to run starfish. To use the container for your analysis, run `docker run -it -v ${path/to/your/data} ${docker_username}/starfish`. The -it flag enables it to interact with the container like a normal shell session and the -v flag allows docker to interact with the supplied directory on the host machine. This is important to enable access to the results after the analysis is finished.l
Operation
Our analysis was performed on Linux Ubuntu 20.04. The starfish workflow required Docker (version 20.10 or later) for containerization. Key tools within the Docker container included bash for script execution, Python 3.8 with the following packages: pandas, matplotlib, and numpy, and Snakemake (version 7.19) as the workflow runner. Additionally, the container relied on pre-installed bioinformatics tools necessary for starfish analysis. Input data and results were managed using the Docker -v flag for directory mounting.
Long read ONT reads are steadily gaining popularity in many metagenomic studies. However, due to platform-based challenges such as high error rare and chimeric artefacts, it is therefore necessary to develop customised bioinformatic tools to effectively characterize microbial composition. We therefore have designed an easy to use workflow to create simulated ONT reads from existing metagenomic studies using ONT (Yang et al. 2017).
Implementation
The workflow implements two distinct steps: simulation and analysis. In the simulation step, the pipeline takes ONT reads from a real metagenome and taxonomically profiles the sample using Lemur and Magnet. More specifically, Lemur first generates relative abundance and taxonomic profiles using a marker gene database and the Expectation-Maximization (EM) algorithm (Sapoval et al. 2024). The profile is then fed into Magnet (“Mimic/README.md at Main · collaborativebioinformatics/Mimic” 2024), which downloads all of the reference genomes and performs competitive read-alignment in order to determine final presence/absence calls. Abundances from Lemur are then mapped to the present genomes called by Magnet to give a final set of species and abundances to use for simulation. The genomes and their abundances are then inputted into Nanosim, along with the number of desired reads to output. Nanosim outputs a simulated file in the FASTA format, as well as error profiles.
The combination of Lemur and Magnet pipelines not only improves recall and precision, but it is easy to deploy as it requires limited computational resources. Apart from simulated reads, Nanosim also generates truth tables built from the simulated reads. These tables contain both taxon labels as well as relative abundance. After simulation, Kraken2 can be run on the simulated reads and the resulting relative abundances are evaluated against the truth table, resulting in precision and recall metrics.
Operation
Mimic is openly available for use at https://github.com/collaborativebioinformatics/Mimic. Mimic has been tested on Linux-based systems and can be run by following installation instructions provided in the repository. The pipeline is implemented in Python and follows the workflow described in Figure 5.
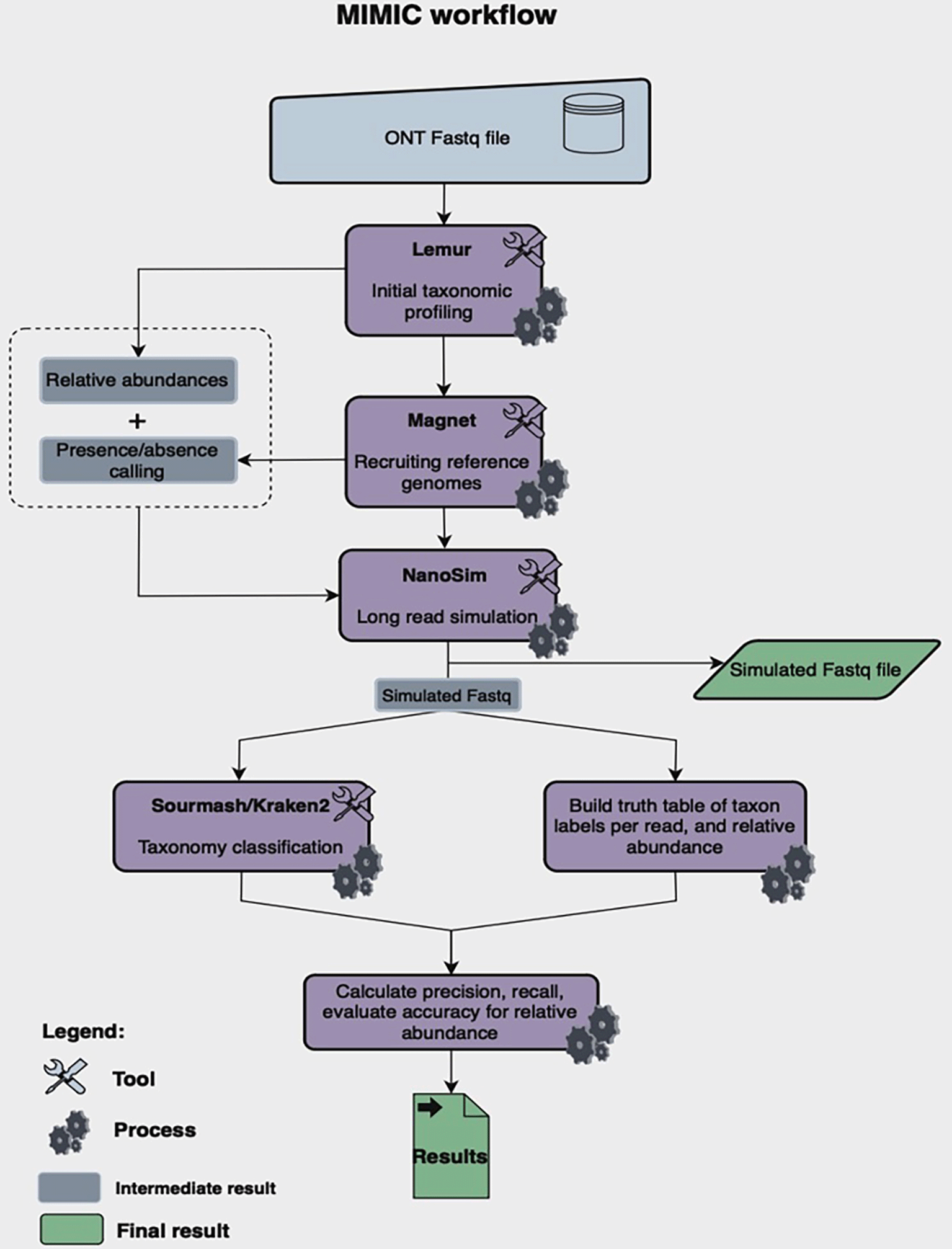
MIMIC simulates Oxford Nanopore (ONT) reads from any existing metagenomic community by 1.) Taking in an ONT FASTQ file and analyzing it with Lemur and Magnet. 2.) Simulating reads based on the Lemur and Magnet mimicked profile with Nanosim. 3.) Running kraken2/sourmash for taxonomic classification and generating truth tables are generated for simulated data based on real microbiome samples and Lemur.
Implementation
In the first step, we downloaded genomic data in the VCF format from 1000Genomes (“A Global Reference for Human Genetic Variation” 2015). Initially, we planned to use VCF files for three populations (Dai Chinese (CDX), Puerto Rican from Puerto Rico (PUR) and British from England and Scotland (GBR)), however, for the purpose of the hackathon, we focused on one population - Chinese Dai in Xishuangbanna, China (CDX) (“Data Portal,” n.d.). The data of the CDX population contained 109 individual samples, the VCF files of which had already been phased with SHAPEIT2 (Delaneau, Zagury, and Marchini 2013), which facilitated our hackathon effort and allowed us to move directly to the next step without phasing haplotypes. Since we planned to use ARG-Needle in the next step, which requires HAP files as input, we used Plink2 to convert the phased VCF files to HAP files (command: `plink2 --vcf phased.vcf --export hap --out new_filename_prefix`) that we subsequently splitted into haplotype blocks, which we defined as parts of the genome between recombination hotspots using the b36 genetic map (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/technical/reference/genetic_map_b36.tar.gz). We acknowledge that we used the old b36 genetic map instead of a new one, however we were not able to run the software with the new hg38 genetic map (https://genome.ucsc.edu/cgi-bin/hgTables) during the hackathon. Therefore, for the purpose of testing the proof-of-concept workflow, we proceeded with the old genetic map, and as a result, we obtained 2089 haplotype blocks for chromosome 6 of the CDX population. Furthermore, we planned to use ARG-Needle (https://github.com/palamaraLab/arg-needle-lib) (Zhang et al., 2023) to infer genealogical relationships between two haplotype blocks - a haplotype block that overlaps with the human leukocyte antigen HLA-A gene (chr6:29631001-30180001) and a random haplotype block (chr6:594001-655001). For that purpose, we used the HAP files corresponding to the first haplotype block of chromosome 6 as input for ARG-Needle (`arg_needle --hap_gz CDX_chr6_HLA.hap.gz --map genetic_map_b36 --chromosome 6 --out CDX_chr6_HLA --mode sequence`). We noticed that the input data did not follow the specifications required by the software (e.g., the genetic map did not contain the required number of sites, even if lifted to the hg38 reference genome using the UCSC Genome Browser (Navarro Gonzalez et al. 2021), however, we successfully obtained the output ARG for a small fraction of the data. As a result, we produced an ARGN file and used the tskit library (https://tskit.dev/tutorials/viz.html; https://github.com/tskit-dev/tskit) in Python to convert the file into a tskit. TreeSequence object for visualization and analysis.
Operation
During the hackathon, we developed a prototype bioinformatic workflow to calculate the similarities between haplotype blocks derived from population genomic data ( Figure 6). The workflow designed and developed during the hackathon includes haplotype phasing, genealogical relationship inference and haplotype block similarity estimation.

We also noticed that the calculations required a lot of computational resources, therefore we performed all calculations on a DNAnexus Cloud Workstation (16 CPUs, 128 GB of memory, 600 GB of storage) (https://documentation.dnanexus.com/developer/cloud-workstation ).
Considering the short timeframe of the hackathon (3 days), we focused our effort only on the proof-of-concept mentioned above, however, we expect that the workflow can be further extended to include the rest of the haplotype blocks of all chromosomes from the CDX population, as well as other populations from 1000Genomes and other large-scale datasets (e.g., GIAB, UK Biobank). In addition, we aimed to build an automated and efficient DNAnexus Workflow (https://documentation.dnanexus.com/developer/workflows) that would take VCF files as input and generate a similarity matrix to compare haplotype blocks.
The pipeline MoVana (MOsaic structural Variants ANnotation in cAncer) is designed to select mosaic events based on their allele frequency (AF), annotate them with overlapping genes and perform the gene set enrichment analysis to infer functional impact ( Figure 7).
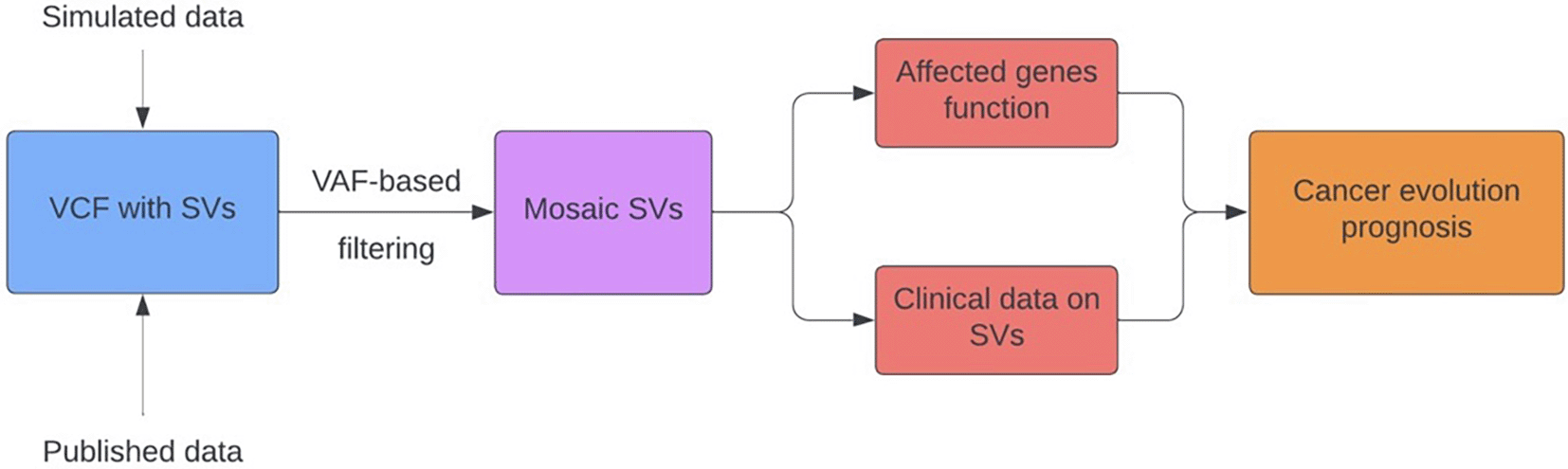
Data
Our workflow involved a publicly available dataset of SV calls from the International Cancer Genome Consortium (J. Zhang et al. 2019). The dataset contains over 71,000 reported deletions, duplications, inversions, and translocations, with the latter excluded for simplicity. To compensate for the fact that the dataset has no estimated AF values, we simulated a distribution of hypothetical AFs. Under a neutral evolutionary model most of the events in a tumor have low AF and belong to the so-called “neutral tail” of the distribution, while true clonal events cluster towards 0.5 AF of heterozygous variants (Hsieh et al. 2020).
Implementation
Our group of interest are subclonal events with lower AFs, therefore the first step of the pipeline involves filtering the dates and including only putatively mosaic events. BCFtools is used to filter out the entries above the user-specified threshold, set to 0.4 in this example. In the next step of the pipeline, known SVs breakpoints and gene coordinates are used to find overlaps with bedtools and annotate each event with the respective affected genes. An extra filtering step is required before the gene enrichment analysis, as the disruptive effect of various rearrangements on gene function depends on the type of SV. Thus, the workflow includes a filtering step based on the SV type, and its output can be submitted for the gene set enrichment analysis or search among known genes implicated in cancer.
Operation
System requirements:
➔ Operating System: Linux (Ubuntu 22.04 or later recommended)
➔ Processor: Multi-core Intel, AMD or ARM CPU
➔ Memory: Minimum 16GB RAM (32GB recommended for larger datasets)
➔ Storage: At least 4GB of free disk space
➔ Software Dependencies:
Workflow overview:
• MoVana workflow
To run the workflow, execute the MoVana_Workflow.wdl file located in the WDL directory using Cromwell, as mentioned on the GitHub page.
Implementation
The project consisted of three parts that constitute a workflow shown in Figure 8:
• Population SV Detection: The pipeline will accurately identify common structural variants (SVs) across multiple datasets, ensuring consistency and reliability in detecting both known and novel variants.
• Phenotype Association: Each identified SV will be linked to phenotypic data such as ClinVar, allowing the correlation of specific genetic variations with particular traits or diseases.
• VCF File Output: The results will be condensed into an annotated variant call format (VCF) file, summarizing the detected SVs and their associated phenotypes. Users can then input a patient ID to retrieve potential phenotypic outcomes based on the identified SVs.
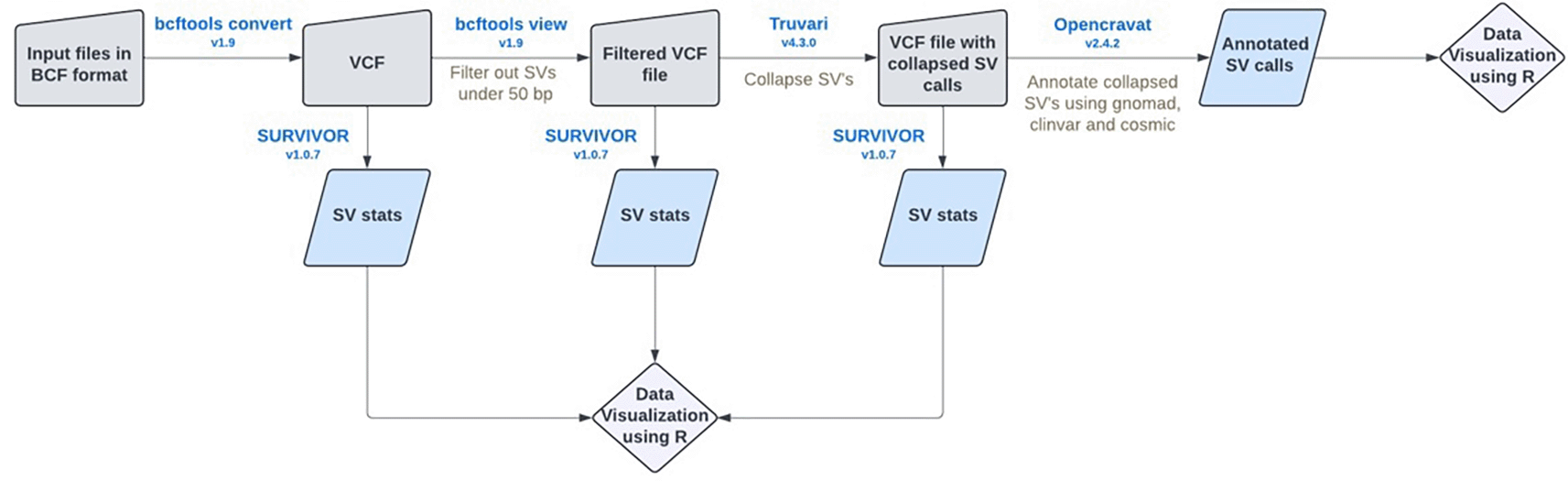
Operation
We gained access to a collection of VCFs created to find Tandem Repeats (TRs) (A. C. English et al. 2024) from a collection of 86 haplotypes accumulated from (Garg et al. 2020; Ebert et al. 2021; Jarvis et al. 2022) and (Wang et al. 2022). To effectively assess relatedness between SVs, we need to set a similarity percentage threshold. For example, if obesity is associated with a 100 bp SV compared to the population reference, we would want to determine if 80 out of the 100 bps (80%) are the same. This is one of the goals of the Truvari (v4.2.2) software (A. C. English et al. 2022), which explains that although similar SVs may be present in different samples, they can occur at different loci along the genome. They also caution that over-filtering with the collapse command may remove important regions of the SV. Additionally, we need to carefully define the range of each SV and consider whether to include, for example, a 5 bp buffer on either side of an SV to account for unique alleles. At this point, we could run SURVIVOR (v1.0.7) (Sedlazeck et al. 2017) for analyzing the VCF data. Finally, we used OpenCRAVAT (v2.8.0) (Pagel et al. 2019) to annotate the VCF file using the ClinVar and gnomAD databases and hg38 genome reference. We removed a problematic line of the VCF header (FILTER/COV) before running the input collapsed SV VCF through OpenCRAVAT. We also added in cosmic, gnomad_gene, clinvar_acmg annotators.
After using OpenCRAVAT to annotate the collapsed SVs, individual structural variants were called to associate a patient with an ontology/diagnosis. At this point, all allele frequencies and structural variant occurrences could be analyzed in R.
System requirements: Laptop for data visualization (in R), HPC cluster for SV clustering and annotation
Operating system: Linux (HPC)
Processors: 32 CPUs (single allocated slurm node on HPC cluster)
Memory: 80 GB RAM (single allocated slurm node on HPC)
Software dependencies: Truvari v4.2.2, SURVIVOR v1.0.7, OpenCRAVAT v2.8.0, bcftools v1.9
Workflow overview:
1. Convert input SV files from bcf to vcf format if necessary using bcftools convert (-O v).
2. Filter out SVs under 50 bp in length using bcftools view (-i “SVLEN>50”).
3. Bgzip compress and index filtered SV VCF using bgzip and tabix.
4. Collapse SVs with truvari keeping the most common variant in each case (-k common).
5. Remove VCF header line that prevents OpenCRAVAT from running using bcftools annotate (-x “FILTER/COV”).
6. Annotate resulting VCF with gnomAD, ClinVar, and COSMIC databases given hg38 reference using OpenCRAVAT (-l hg38 -a gnomad gnomad_gene clinvar clinvar_acmg cosmic cosmic_gene -t text excel).
GTF annotation
We used the reference GTF annotation file from GENCODE (v46) to annotate the TR loci in our database. This annotation provides information on whether a particular TR region is located within an exon, a gene, or in intergenic space. By adding this information, we were able to analyze the varying strengths of TRs across different regions and to assess their impact on population structure prediction.
Length polymorphism score
To calculate the length polymorphism score, we first assessed the allele frequencies and allele counts of individual TRs for each ancestry group separately. The length polymorphism score is a per-locus measure of the proportion of distinct alleles by length relative to the total number of alleles measured at that locus. For the length polymorphism we used a query as tdb query len_poly_score hprc_105.tdb > result.txt.
Fixation index (Fst)
Fixation index (Fst) is a measure of genetic differentiation between populations, quantifying the proportion of genetic variance due to population structure (Meirmans and Hedrick 2011). Here we calculated the Fst of TR alleles across loci. We first run a query to calculate allele counts by population using population_ac_by_length.py to create input_allele_counts.tsv using an equation from (Sampson et al. 2011). We made a query on Fst using python calculate_fst.py -o result.tsv input_allele_counts.tsv.
Population informative TR loci
A baseline PCA of TR alleles across 105 samples showed a decent clustering regarding super population structures ( Figure 9). In the next step, by filtering all TR loci based on our conditions (fixation index > 20 and length polymorphism score > 20), we identified 14 loci of interest for further investigation: 7 within genes, 6 in intergenic regions, and 1 in an exon. We performed PCAs on the loci in genes and intergenic regions to explore the role of TRs in these regions in predicting population structure.

We selected chromosome 22 from the HG002 BAM file to test the TykeVar pipeline due to its high coverage (130x), which ensures reliable detection of variants and provides an ideal dataset for simulating mosaic alterations. Using the TykeVar pipeline, we introduced artificial variants into the BAM file to mimic a range of variant scenarios relevant to mosaicism. Figure 10 (A) illustrates the process of incorporating insertion variants into the BAM file, while Figure 10 (B) showcases mosaic deletions introduced exclusively in the modified BAM file, which were absent in the original dataset. To assess the performance of TykeVar, we employed the Sniffles2 mosaic variant caller, achieving over 80% accuracy in detecting the artificially introduced mosaic reads. These findings confirm that the TykeVar pipeline effectively generates realistic mosaic variants and that our detection strategy is robust, validating the approach for simulating and identifying specific genomic alterations in modified BAM files.
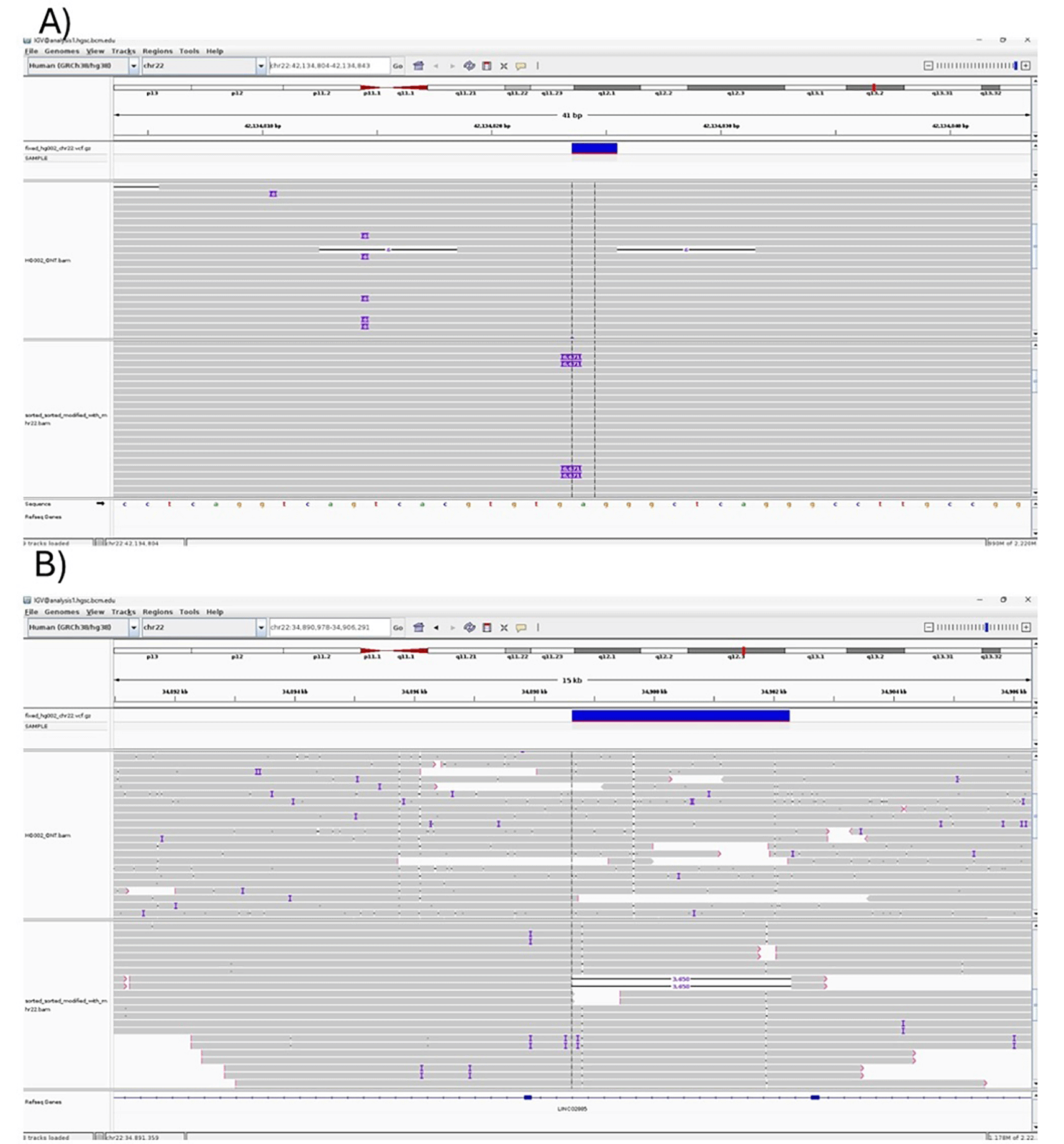
(A) Injection of artificial variants into the HG002 chromosome 22 BAM file using the TykeVar pipeline.
(B) Visualization of deletions in the modified HG002 chromosome 22 BAM file after TykeVar pipeline processing.
We generated overall statistics, temporal analysis and visualization of spatial distribution. Our most analysis focused on four most abundant pathogenic species including Pseudomonas aeruginosa, Acinetobacter baumannii, Klebsiella pneumoniae, Escherichia coli.
Most organisms contribute few AMR genes to the database, while few organisms contribute the bulk of AMR genes. Regarding the number of AMR genes of one species, Pseudonomas aeruginosa, Acinetobacter baumannii, Klebsiella pneumoniae and Escherichia coli far outweighed other species while some clinically significant pathogens did not rank high. For instance, Streptococcus pneumoniae. Most targets of AMR genes included penicillins (penams), carbapenems, monobactams, and cephalosporins, which are all beta-lactams, i.e., the antibiotics inhibiting the synthesis of bacterial cell walls. Other significant targets were aminoglycosides (e.g., amikacin), tetracyclines (e.g., doxycycline & tetracycline), and peptide antibiotics (e.g., bacitracin). The most prevalent antibiotic mechanism is “antibiotic inactivation”, while “antibiotic target alteration” and “antibiotic efflux” ranked second and third respectively.
Our temporal analysis focused on four top species. The occurrence of resistance increased by time in all countries and all types of antibiotics. However, this needs to be normalized by the number of samples in each year because the amount of recent data is greater than early years. If we looked at the mechanism of resistance, the prevalence of each mechanism fluctuated mildly in each species. “Antibiotic efflux” was the most common mechanism in three species but K. pneumoniae had “antibiotic target alteration” the most ( Figure 11).
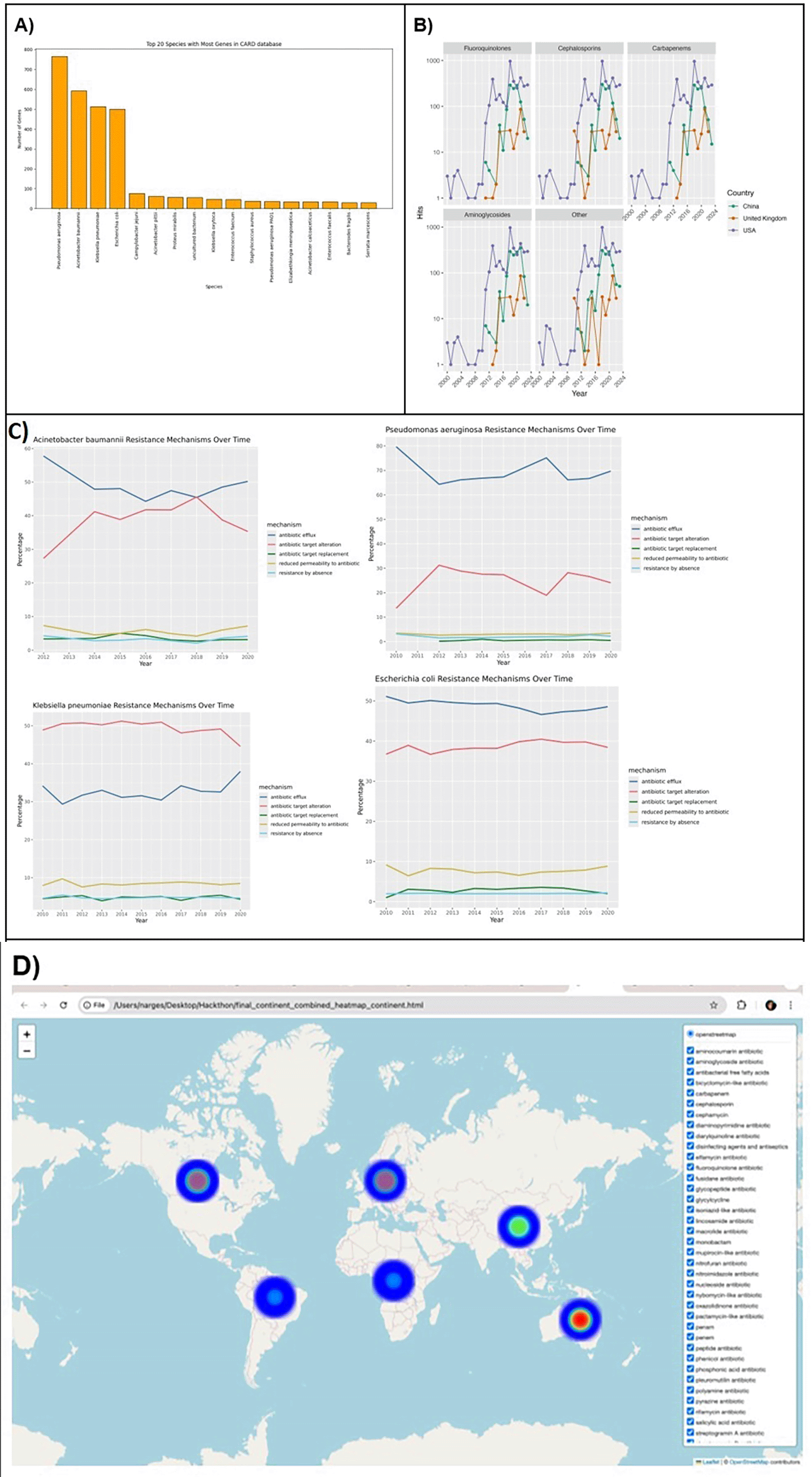
As a result of the hackathon, starfish is now available as a docker container and snakemake pipeline. The use of containerized environments (e.g., docker) and workflow management systems (e.g., snakemake) is crucial for ensuring the reliability and reproducibility of bioinformatics analyses. The docker container provides a consistent and isolated environment, encapsulating all the necessary software dependencies, libraries, and configurations needed to run analyses. The snakemake pipeline enables the automation and organization of the starfish analysis, ensuring that each step is executed in a specified sequence with minimal human intervention. It also enhances the reliability of analyses by allowing for error tracking, version control, and easy debugging. Furthermore, both Docker and Snakemake enable comprehensive documentation of the analysis pipeline, making it easier for others to understand, validate, and reuse the methods. Together, these two tools improve the accessibility and usability of starfish, to facilitate its application to non-fungal genomes.
We ran the Mimic pipeline on the following SRA samples (SRR29660113 and SRR30413550). Metagenomes were simulated at 1k, 50k, and 100k reads for both ‘perfect’ reads and default error-prone reads, which in this case reflected a ~11% sequencing error rate (4% mismatch, 4% insertion, 3% deletion), which is high but not unrealistic for current ONT devices. For the 1k reads we evaluated the accuracy of Kraken2’s classification of each read at the genus level and above for SRR30414550. Here, the “FN” counts reflect reads that are not classified by Kraken2 at that taxonomic rank. For the ‘perfect’ reads, we get:
rank FN TP FP TN Prec Rec
genus 47 799 154 0 0.838 0.799
family 41 862 97 0 0.899 0.862
order 39 868 93 0 0.903 0.868
class 33 955 12 0 0.988 0.955
phylum 29 965 6 0 0.994 0.965
While for the error-prone reads, we get:
rank FN TP FP TN Prec Rec
genus 227 566 206 1 0.733 0.567
family 219 589 191 1 0.755 0.590
order 214 599 186 1 0.763 0.600
class 176 792 31 1 0.962 0.793
phylum 171 798 30 1 0.964 0.799
These preliminary results illustrate the potential impact of sequencing errors on the accuracy of Kraken2 taxonomic classifications. Kraken2 performs well at all taxonomic ranks for error-free reads. However, when sequencing errors are introduced, Kraken2’s precision and recall drop quickly, particularly at lower taxonomic levels. It is important to note that Kraken2 was designed for use with short, accurate reads and so all considered, demonstrates good flexibility when being used on these long reads.
Although we were able to successfully demonstrate MIMIC’s ability to generate and evaluate simulated ONT reads, we were not able to deploy it onto the DNAnexus environment. However, we were able to detect the reduced precision of Kraken2 performance on reads with simulated error. The next step would be to benchmark our tool against existing metagenomic long read simulators such as CAMISIM. Furthermore, by adding gene gain/loss events in the reference genomes, this can aid in simulated datasets with known ‘ground truth’, that can be either evaluated using existing tools or build efficient pipelines that can effectively quantify these variations.
During the hackathon, we used the ARG-Needle software to produce ancestral recombination graphs (ARGs), which are collections of trees that contain nodes corresponding to individual genomes and their ancestors, and edges representing the evolutionary inheritance of genomic variants, for the Chinese Dai in Xishuangbanna, China (CDX) population from the 1000Genomes Database. We hypothesized that we could analyze intra- and interpopulation genomic variation in specific regions of interest (e.g., immunological genes, such as HLA-A) by comparing haplotype blocks that overlap with those regions, therefore, we developed a workflow for converting haplotype blocks into similarity matrices. We expected that such similarity matrices could be useful for studies that examine how recombination affects the genomic structure of a population, or how cis- and trans-effects impact the rare variant penetrance.
We produced a proof-of-concept ARG for a small fraction of the haplotype block (chr6:136011-160001) in the Chinese Dai in Xishuangbanna, China (CDX) population from 1000Genomes. To analyze the result, we converted the ARGN file corresponding to the ARG into a tskit. TreeSequence object, and used tskit to summarize the results ( Figure 12) and to visualize the first tree of the ARG ( Figure 13).
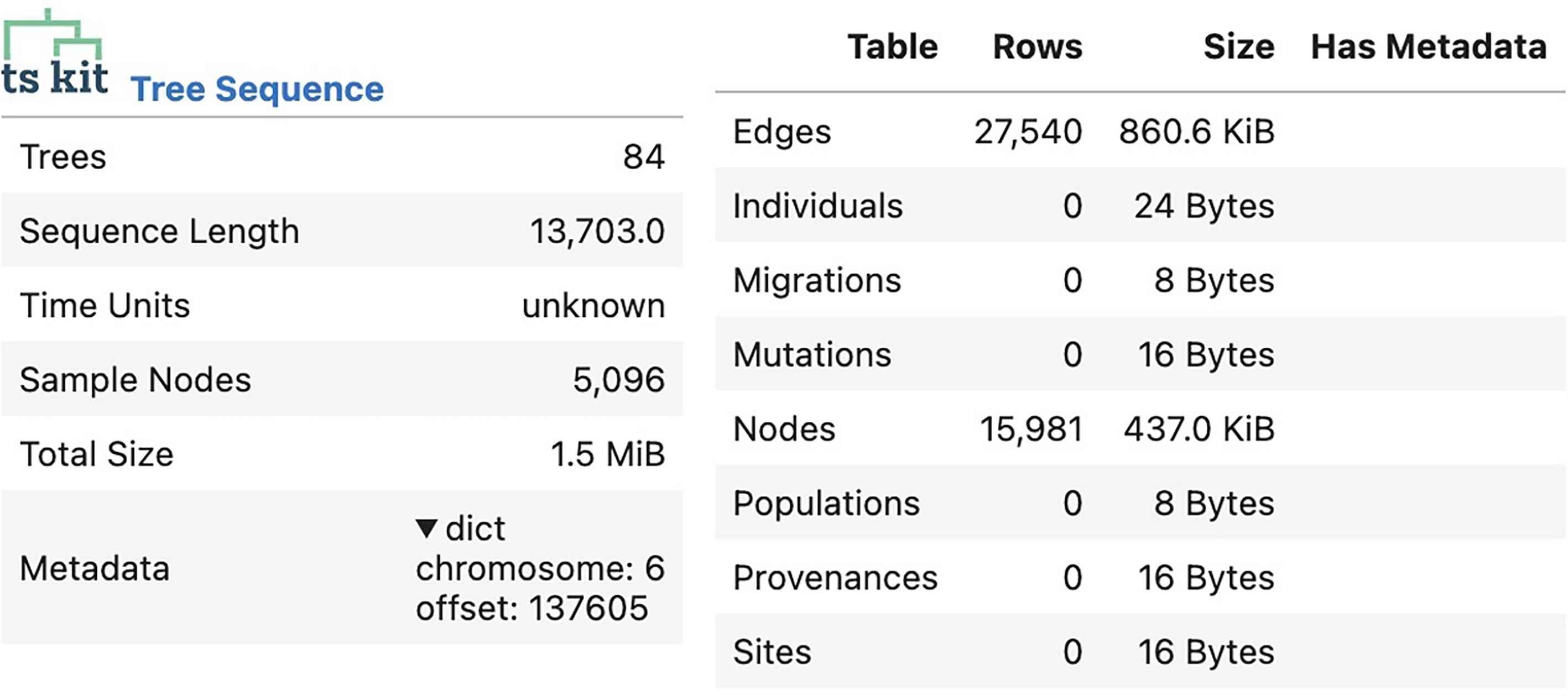
The ARG contains 84 individual trees with 15,981 nodes and 27,540 edges.
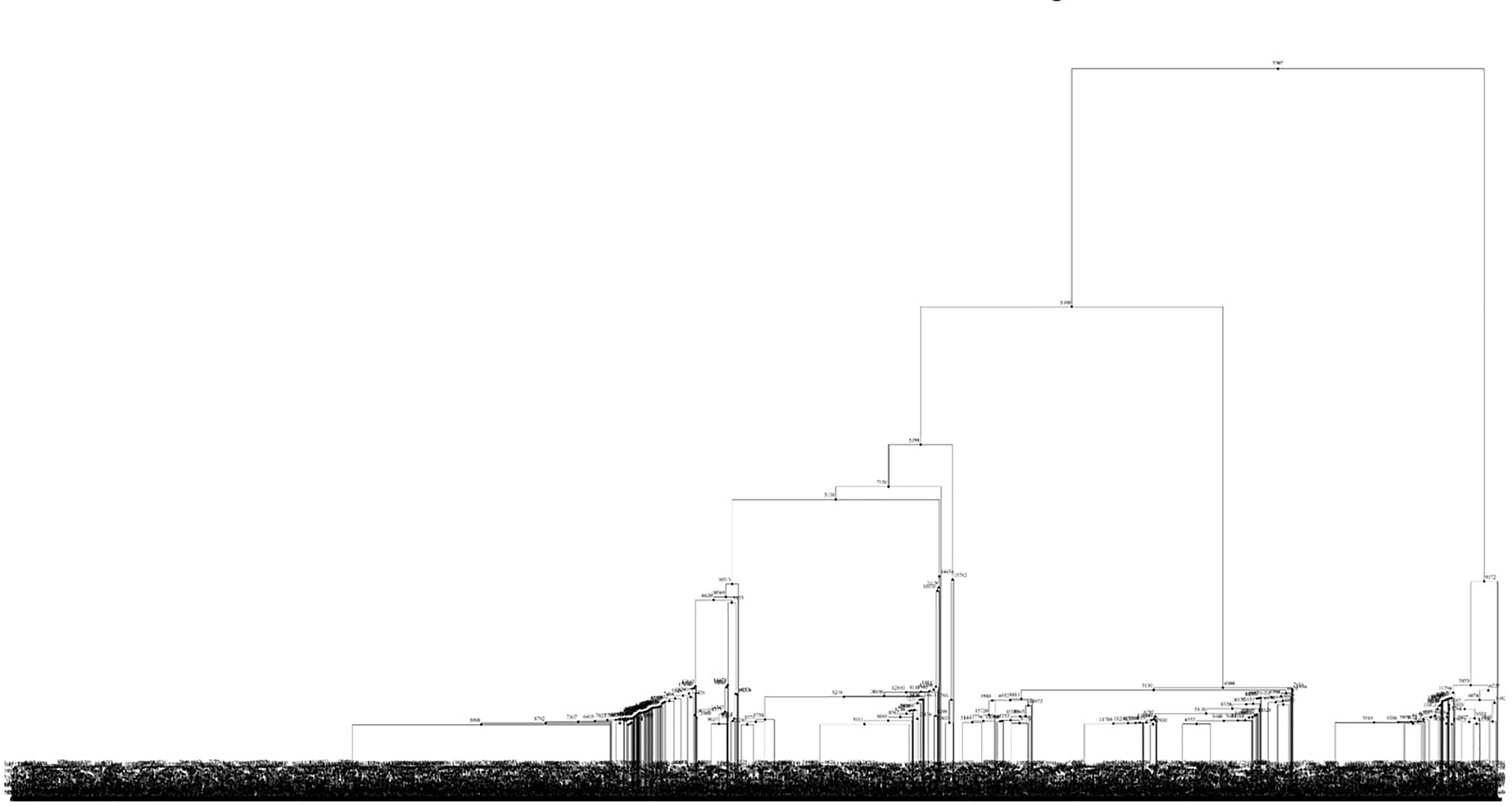
Each tree of the ARG represents a fraction of the genome in the CDX population that shares common ancestry. The first tree that we analyzed shows how genomic variation associated with recombination events in one genomic region has been inherited within the population. Further analysis of the ARG, as well as a comparison to the ARG of the haplotype blocks overlapping with other regions of interest could reveal individuals’ risk for certain diseases or inheritance patterns in polygenic diseases, however it was beyond the scope of this hackathon project to conduct such analyses.
Overall, the results of this project constituted an exploration of the idea to analyze haplotype blocks in such a computationally efficient and inexpensive way.
We acknowledge that this undertaking was fraught with challenges inherent to the hackathon framework, such as a lack of time for data exploration and preprocessing, as well as technical difficulties with running the software (Busby et al. 2016). Nevertheless, we expect the result of our project to be useful for the scientific community and serve as a future reference for further projects.
In our mock example, we applied the tool to a subset of reported structural variants (SVs) from the International Cancer Genome Consortium. After filtering for variant allele frequency (VAF), the dataset included approximately 1,000 deletions, duplications, and inversions. We found that over 900 genes were affected by duplications alone, many of which are involved in known cancer-related pathways ( Figure 14).
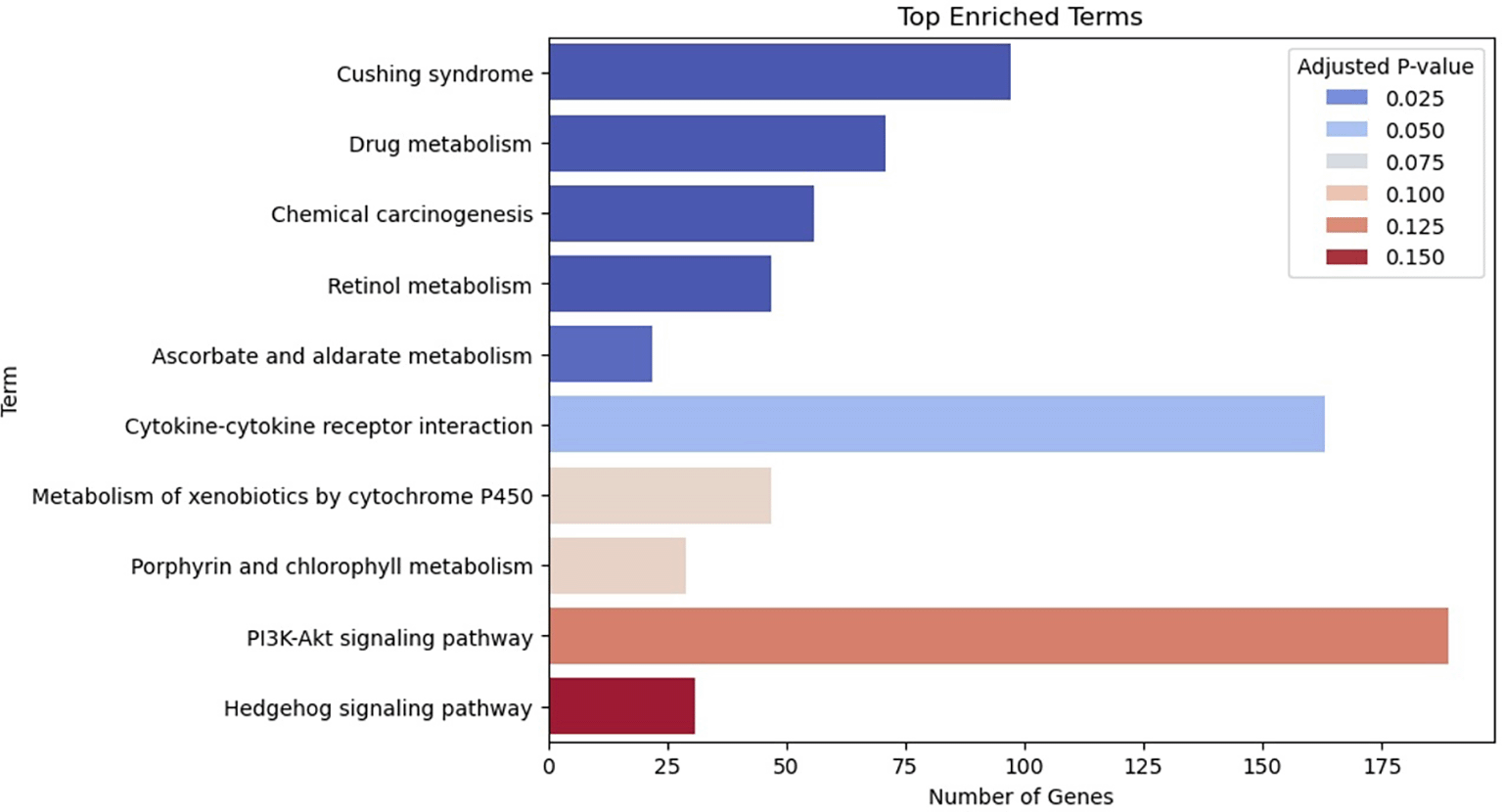
A previous similar study identified 11 SV loci associated with an increased risk for obesity, with an Odds Ratio exceeding 25% (Walters et al. 2013). This project aims to build upon such findings by extending the analysis to a broader set of SVs and phenotypes, facilitating the discovery of novel genetic contributors to complex traits.
Validation
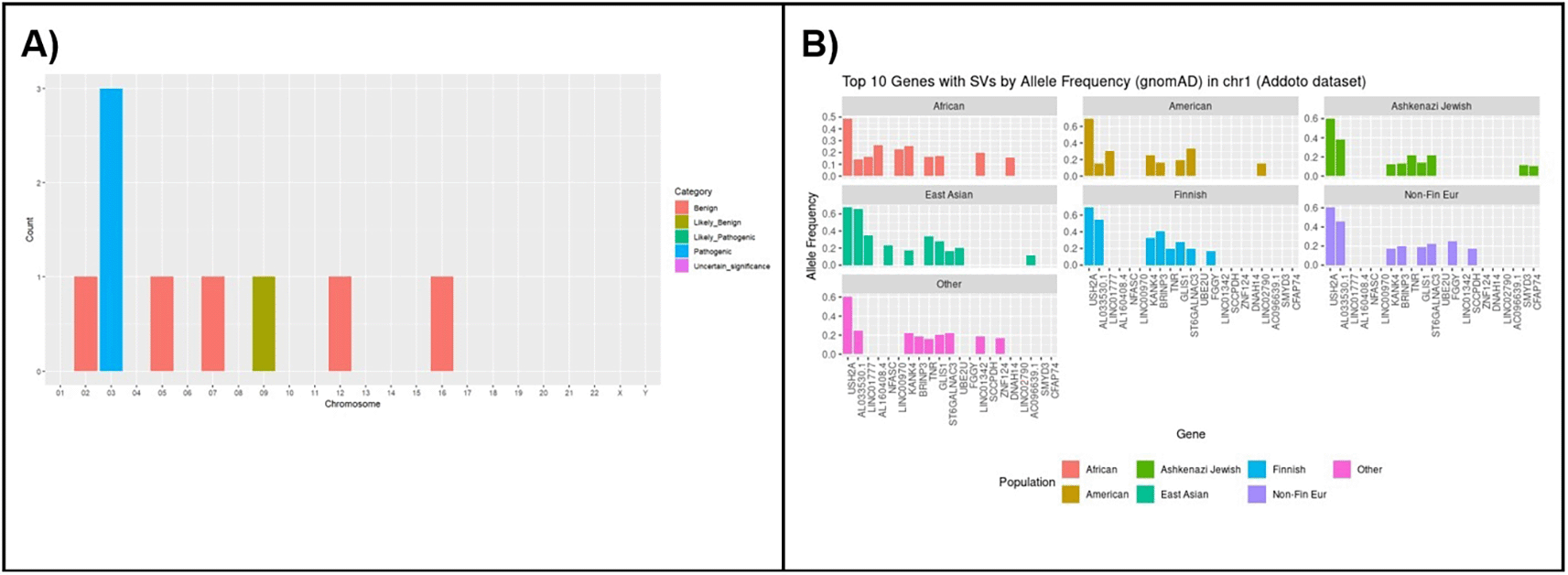
We validated our pipeline on the Project Adotto assembly-based variant calls from the GIAB tandem repeat benchmark (https://zenodo.org/records/6975244), beginning with SV calls in chromosome 1 (either insertions or deletions). Upon SV filtering steps, we went from 194,098 SVs to 55,905 SVs (remove those under 50 bp in length) and then 29,026 SVs (truvari collapse function keeping most common allele in each cluster).
Gene analysis of chromosome 1
In our analysis of chromosome 1 using the Adotto dataset, we identified genes with the most prevalent allele frequencies across different populations. These allele frequencies, including those for structural variants, were sourced from the gnomAD dataset. This analysis highlights genes that show significant variation in allele frequencies among American, Ashkenazi Jewish, East Asian, Finnish, Non-Fin European, and Other populations.
For example, the gene NFASC, which is involved in neurodevelopmental disorders with central and peripheral motor dysfunction (MIM 609145), shows notable structural variants in East Asian ancestry. The prevalence of structural variants of NFASC in this population underscores the importance of understanding population-specific genetic variations, which can inform physicians and researchers about potential genetic risk factors and guide future studies. Our tool with continued research into these population-specific variants is essential for advancing personalized medicine and improving genetic counseling.
Annotating SVs with ClinVar annotations
We have successfully validated our pipeline by gathering all structural variants (SVs) from the Adotto database and combining them with ClinVar data. This analysis led to the identification of three structural variants classified as pathogenic in ClinVar, affecting a total of eight individuals across the dataset ( Figure 15).
To facilitate the use of this information, we have developed an additional tool that converts the data into a user-friendly PDF output. This PDF includes the sample name of each individual and the predicted diagnosis based on the known ClinVar phenotypes. It provides a comprehensive report detailing each variant, including all relevant information. We were able to find individual patients with specific ontologies from associated SVs and their genes. An example of this PDF report is shown in Figure 16.

The location of interest is then related to specific ontologies listed on the ClinVar database.
The concepts developed over the 2024 Baylor College of Medicine/DNAnexus hackathon described here represent novel work across multiple important fields of computational biology. These projects encompass complex regions and variants of the human genome to comprehensive analysis methodologies for AMR across bacteria. These projects individually represent important milestones in their individual fields pushing our capabilities to obtain novel insights into complex data sets and enabling a deeper understanding of important mechanisms. This was enabled by a multinational team of 48 scientists spanning the entire world to facilitate this progress in a FAIR-compliant manner.
A comprehensive single report of all of these measures would further assist researchers in prioritizing tandem repeats. This study will help to subset TRs and further expand TR-specific kinship analysis. This research could be expanded to relate population structure with methylation data as well as compare TR in genes/promoter vs intergenic as well as check sex chromosome vs autosomes. All the codes and scripts for tandem repeats queries have been added to the tandem repeats github repository.
The next steps of our project will focus on developing and releasing a Dockerized version of our mosaic variant detection framework to ensure easy deployment across different environments to improve accessibility and reproducibility. We will further improve the detection of mosaic variants from short-read sequencing data, with a particular focus on identifying single nucleotide substitutions (SNS) and small insertions and deletions (indels) across a range of variant allele frequencies (VAFs). This enhancement aims to increase sensitivity and accuracy in detecting these subtle genomic alterations. Additionally, we will refine the re-genotyping process in the SpikeVar pipeline to improve accuracy in generating the ground truth set for the VCF file from the SpikeVar pipeline.
Currently, our interactive heat maps provide easy visualization of AMR genes and its information on the world map. It is novel but requires more finetunes, including but not limited to, making the interface more user-friendly, some subset options and public accessibility. We concluded some trends across years but it would be more clear after normalization and data cleaning.
This is the first time of deep diving in the entire SRA dataset. With our alignments results and parsed metadate, we can investigate more on association between AMR genes and the environments or the hosts. Phylogenetic analysis is another important aspect. We can study the relationships of similar AMR genes of different species, trace the origin of an AMR gene or a specific resistant strain. Because the long-read sequencing technologies are rising, our dataset provides a good chance to see how the sequencing platforms influence the property of Longan unitigs and the alignment.
This project will not only contribute to the understanding of AMR gene distribution but also provide participants with hands-on experience in handling large-scale genomic datasets and applying bioinformatics tools in a real-world context.
The Docker container has been created for enhanced scalability and reproducibility. Future goals include adding a workflow (e.g., Snakemake pipeline) and the application of Starfish to non-fungal genomes (particularly mammalian). However, there are anticipated challenges of acquiring the appropriate annotation input files and computational time when moving from small fungal to large mammalian genomes. An alternative approach could involve using different computational tools to identify transposons in eukaryotic organisms.
We built MIMIC, a metagenome simulator that creates ONT reads based off of the taxonomic composition and error profile of real metagenomic samples. We showed that we can generate simulated samples that accurately reproduce the conditions of actual metagenomic samples, and that the commonly used taxonomic classifier Kraken2 performs poorly on error-prone long reads. Moving forward, MIMIC provides a simple framework to comprehensively evaluate long-read taxonomic profilers on any sample type, allowing researchers to test or develop tools for more precise real world applications.
While this challenge was difficult for a hackathon, we were able to lay the groundwork for other teams to work on this particular problem. In fact, a team at the Nucleate Hackathon Challenge in Pittsburgh (October 2024; https://www.nucleate.xyz) was able to make some additional headway on this problem: https://github.com/ShijieTang/BioHack_Haplotype, moving away from explicit ancestral recombination graphs. Ancestral recombination graphs offer a promising alternative to study complex genealogical relationships (Lewanski, Grundler, and Bradburd 2023) and moving toward brute force analysis of local haplotype blocks. This work will continue at the Carnegie Mellon University Libraries Hackathon in March, 2025. Please check https://biohackathons.github.io for additional details.
The MoVana pipeline is introduced as a specialized tool to focus on mosaic variants, as opposed to a comprehensive characterization of all events in a given call set. By implementing variant allele frequency (VAF)-based filtering, the pipeline enables the selection of putative subclonal mutations. The workflow then intersects these mutations with affected coding sequences and ultimately identifies all genes in the dataset that overlap with a specific SV type. This approach aids users in linking structural variants to their functional consequences, particularly by identifying pathways impacted by SVs through the gene set enrichment analysis implemented in MoVana.
The future direction of the project includes analyzing mosaic SVs in primary tumors against metastasis and relapse states and finding recurrent mosaic events implicated in treatment resistance. This can be achieved through the integration of multiple variant databases like dbVar, ClinVar, and OncoDB to enhance the accuracy and reliability of clinical outcomes given by the pipeline.
The development of this pipeline represents a significant advancement in the annotation and association of structural variants (SVs) with disorders. By combining gnomAD allele frequencies and ClinVar clinical data, our tool facilitates a more straightforward and efficient approach to detecting and analyzing SVs in patient sequences. The integration of phenotypic information with clinical and larger dataset sources enhances the tool’s utility in patient care, leading to more informed predictions and better clinical decision-making. Sveedy streamlines the interpretation of SV data and enhances the ease of accessing detailed diagnostic information, making it an invaluable resource for clinical research and patient care.
Future Directions: An improvement to Sveedy would include incorporating additional methodologies and databases within the workflow to enhance the accuracy and scope of SV detection and annotation. The tool could also be streamlined by organizing a Binder environment for global accessibility and improving the pipeline in terms of data formatting for efficient SV processing and reduced computational overhead. The streamlined design and future expansions aim to set a new standard for bioinformatics workflows in precision medicine.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)