Keywords
Melophlus sarasinorum, sponge-associated Streptomyces, de novo assembly, whole genome, L-TIR
This article is included in the Genomics and Genetics gateway.
Melophlus sarasinorum, sponge-associated Streptomyces, de novo assembly, whole genome, L-TIR
The version 3 include more information in the methods section and results of generated sequencing data
See the authors' detailed response to the review by Matsapume Detcharoen
See the authors' detailed response to the review by Arzu Karahan
Streptomycetes represent a group of metabolically diverse bacteria known for their ability to produce natural products of considerable importance in human health, veterinary medicine, and agriculture. The genus Streptomyces primarily synthesises bioactive secondary metabolites that include antibiotics, as well as antifungal, antiviral, and anthelmintic agents, anticancer drugs, immunosuppressants, and herbicides.1 However, Streptomyces spp. demonstrate notable GC content exceeding 70% and show high repetition within secondary metabolite biosynthetic gene clusters (BGCs).2 These provide issues in sequencing and high-quality genome assembly for in-depth genome mining, which are presently addressed through intensive sequencing efforts. Advances in genome sequencing technology have uncovered a multitude of hitherto unexamined metabolic capabilities inside Streptomyces genomes. The advent of fast and efficient genome sequencing tools has allowed for the examination of more profound and comprehensive bacterial genomic data. Thus, the data rely on the thoroughly curated genome sequence characterised by high contiguity and improved annotation. In recent years, the emergence of long-read sequencing technologies has led to the integration of both Illumina (short) and Nanopore or PacBio (long) reads, potentially enhancing the completeness of genome assemblies relative to those constructed based solely on Illumina reads.3
Streptomyces sp. BTA 1-131 was isolated from an Indonesian sponge, namely MS5, identified as Melophlus sarasinorum. The marine sponge M. sarasinorum is widely distributed in the Indo-Pacific region, including Indonesia, Palau, Guam, Papua New Guinea, and the Solomon Islands.4 A multiomics analysis of M. sarasinorum leads to the exploration of the previously unknown natural products derived from the sponge M. sarasinorum and the bacterial symbiont.5 Indeed, our initial study showed the potential extracts of sponge M. sarasinorum MS5 to inhibit the growth of gram-positive pathogenic bacteria, and the abundance of actinobacteria is significantly correlated to the sponge bioactivity.6 Further investigation observed antibacterial activity in the methanolic extracts of the sponge-associated actinobacterium Streptomyces sp. BTA 1-131 against the gram-positive S. aureus ATCC 13240. Additionally, the methanolic extracts of Streptomyces sp. BTA 1-131 also demonstrated cytotoxic effects on the MDA-MDB231 human breast cancer cells and the Caco-2 colorectal adenocarcinoma cell.7 The study confirmed that potential bioactivity was present between the sponge-associated actinobacterium Streptomyces and its sponge host.
Molecular identification using the 16S rRNA gene sequence revealed that the strain BTA 1-131 is 98% similar to S. kunmingensis NBRC 14463.7 A study by Wei et al. (2017)8 reported the potential of S. kunmingensis YIM 121234, which originated from soil, to produce bioactive compounds with potent cytotoxic activity towards the MCF-7 human breast cancer cells. Moreover, another reported strain, S. kunmingensis MR03, originated from marine sediment and showed its potential to synthesise cadmium sulphide nanoparticles that were active against biofilm-forming bacteria such as S. aureus, E. coli, P. aeruginosa, and Enterecoccus spp.9
The diversity of Streptomyces and their novel compounds originating from marine biospheres remains less explored.10 Indeed, the analysis of secondary metabolite BGCs demonstrated by Almeida et al. (2019)11 suggests that while the marine isolates share a close genomic relationship with their terrestrial counterparts, they may have the capacity to produce distinct compounds. Considering the atypical origin of the culturable sponge symbiont Streptomyces sp. BTA 1-131, similarity of the strain to known Streptomyces (≤ 98.7%), and our intention to further evaluate its biosynthetic capabilities, we sequenced the whole genome of Streptomyces sp. BTA 1-131, using a de novo approach based on the hybrid Illumina and Nanopore platform. This has allowed us to generate a complete chromosome sequence. To the best of our knowledge, this is the first study to publish the complete genome of a sponge-associated Streptomyces strain from the marine sponge M. sarasinorum of Indonesian origin. The reported data provides the basis for further analysis to explore the strain as a putative new Streptomyces species and reveals the biosynthetic capabilities of its bioactive compounds.
Streptomyces sp. BTA 1-131 was previously isolated from marine sponge M. sarasinorum in December 2015. The sponge was collected in Tanjung Batu Angus, Lembeh Strait, Bitung, North Sulawesi, Indonesia (1.50572° N, 125.24541° E; depth 13 m) as described in the previous study.6 The strain BTA 1-131 was previously identified by sequencing the 16S rRNA gene and is closely related to soil-derived S. kunmingensis NBRC 14463.7 The nucleotide sequence of the 16S rRNA was deposited in the NCBI GenBank database under accession number MT280129, and the cultivable isolate was deposited in the Indonesian Culture Collection BRIN (InaCC) under accession number A1205.7 In this study, the strain BTA 1-131 was revived from a glycerol stock stored at –80 °C. Prior to genomic DNA extraction, the strain BTA 1-131 was grown on an International Streptomyces Project (ISP)-recommended medium, ISP2 agar medium, at 28 °C, for 14 days. The ISP2 medium contains yeast extract (BD Difco, USA; 4 g L−1), malt extract (BD Difco, USA; 10 g L−1), and dextrose (BD Difco, USA; 4 g L−1). The medium was prepared in 1 L of seawater with an addition of bacteriological agar (BD Difco, USA; 20 g L−1).
Genomic DNA was extracted from Streptomyces sp. BTA 1-131 colonies grown on ISP2 medium agar plate using the Quick-DNATM HMW MagBead kit according to the manufacturer’s instructions (Zymoresearch, USA). The integrity of the extracted genomic DNA was assessed by gel electrophoresis (0.8% tris borate-EDTA agarose w/v), while its quality and quantity were analysed by a NanoDrop spectrophotometer (Thermo Fischer Scientific, USA) and a Qubit 4 fluorometer (Thermo Fischer Scientific, USA) using the QubitTM dsDNA High Sensitivity assay kit (Thermo Fischer Scientific, USA), respectively. Moreover, total gDNA was used for the input of library preparation for both short read-based Illumina sequencing and long read-based Oxford Nanopore Technologies (ONT) sequencing platforms.
To sequence the genome of the strain BTA 1-131 on the Illumina platform, the DNA library was prepared using the Illumina NextSeq500 system with the NextSeq 500/550 kit v2.5 (300 cycles) to generate 2 x 150 bp paired-end sequence reads following the manufacturer’s protocol (Illumina, USA). For the ONT platform, the DNA library was prepared for GridION ONT using the SQK-NBD114.24 ligation sequencing kit according to the manufacturer’s recommendation (Oxford Nanopore Technology, UK). Kit from NEBNext, FFPE Repair Mix (NEB M6630, UK) and Ultra II End repair module (NEB E7546, UK) were employed in the library preparation. In brief, total gDNA was repaired using an end-prep enzyme mix (NEB M6630 & NEB E7546), generating DNA with 5’ phosphorylated, 3’ dA-tailed ends. Barcodes were ligated with an ONT-compatible adapter. The library was quantified with a Qubit Fluorometer before loading to the FLO-MIN114 flow cell. Sequencing control was conducted in MINKNOW v23.04.6 following default sequencing run from ONT (72 h). The output raw reads were set to POD5 for a separate basecalling process.
Illumina read processing
Illumina-generated FASTQ files were initially processed using Fastp v0.24.012 to assess read quality and remove low-quality bases (Data file 1).13 Reads were filtered with the parameters -q 30 -l 100 -c, ensuring only high-quality reads were retained (Data file 2).14 Unpaired reads (those lacking a mate from either R1 or R2) were kept using the flags –unpaired1 sample_unpaired1.fastq.gz --unpaired2 sample_unpaired2.fastq.gz and were included alongside paired reads for subsequent hybrid assembly with the ONT data.
ONT read processing and hybrid assembly
Raw POD5 files were basecalled at super accuracy using Dorado v0.8.2, generating a simplex BAM file. This BAM file was converted to FASTQ format using Samtools v1.21.15 Read quality distributions were assessed with NanoPlot v1.44.0,16 (Data file 1).13
Hybrid assembly began by assembling ONT reads first using Flye v2.9.5,17 (Data file 3).18 Parameters included --nano-hq -g 8m --asm-coverage 50, reflecting an estimated 8 Mbp genome size and a coverage limit set to 50X. Assembly graphs were visualised with Bandage v0.9.0,19 (Data file 4).20 Moreover, to refine the ONT-based assembly, Medaka v2.0.1 (Medaka consensus) was applied with recommended settings optimised for v14 reagent kit reads. A final hybrid assembly was then generated by polishing the ONT assembly with the high-quality Illumina reads using Pilon v1.24.21 Three iterative polishing steps were performed; in the first iteration, the Illumina was aligned to the Medaka-polished ONT assembly using Bowtie v2.5.4,22 and alignments were processed with Samtools v1.21. The resulting consensus contigs become assembly sources for the second iteration and continue until three iterations of polishing are reached. After these three polishing rounds constituted the final hybrid assembly for subsequent analyses.
Assembly statistics and completeness
Assembly metrics were obtained with the quality assessment tool for genome assembly (QUAST) v5.2.0,23 reporting the number of contigs, N50, and GC content; gene predictions were disabled for this step (Data file 5).24 Genome completeness was evaluated using the benchmarking universal single-copy orthologs (BUSCO) v5.8.225 in genome mode, employing the Streptomyces_odb12 lineage dataset (Data file 6).26 The identified single-copy orthologs were visualised via the generate_plot.py script included with BUSCO. CheckM v1.2.327 was used with the lineage_wf workflow to estimate completeness and contamination, summarised in a table of reported statistics (qa) (Data file 7).28 Assemblies with >95% completeness and <5% contamination were considered high-quality.
Genome visualisation and annotation
Genome visualisation was performed using GenoVi v02.16,29 which generated circular genome maps depicting sequence features, contig boundaries, and genomic architecture. Functional annotation of clusters of orthologous groups (COGs) categories was conducted using the integrated DeepNOG30 within GenoVi, with gene sequences queried against the COG database to assign functional categories and identify orthologous proteins across genomes (Data file 8).31 The resulting visualisations provided both an overview of overall genomic organisation and detailed functional annotation profiles for downstream analysis.
TIR and plasmid identification
To examine the linear inverted repeats on the strain BTA 1-131 chromosome, a basic local alignment search tool (BLAST) was conducted using the parameters described previously by Jørgensen et al. (2024).32 While putative plasmid contigs were identified with PLASme v1.133 using the default database and the “balance” mode parameter. All other settings were kept at default values. Any candidate plasmid contigs were verified by examining the assembly graph in Bandage to confirm their circular topology or unique structural features.
Taxonomic inference and phylogenomic analysis
Taxonomic assignment was carried out on the type (strain) genome server (TYGS)34 by submitting the polished genome assembly (Data file 9).35 Additional inferences were made using GTBD-Tk v2.1.0,36 incorporating Streptomyces kunmingensis strains and the type strain S. albus NRRL B-1811 (ASM72588v1), as well as some selected Streptomyces strains associated with marine sponges reported by Xu et al. (2019)37 for comparative analysis. Moreover, Deferribacter desulfuricans SSM1 was chosen as an outgroup. The resulting genome BLAST distance phylogeny (GBDP) tree was visualised in iToL v7.38 The TYGS webserver also provided a DNA-DNA hybridization (dDDH) score.
To assess the genomic similarity between the polished genome assembly of the strain BTA 1-131 and one of the reference S. kunmingensis DSM 41681 (the nearest strain based on the genome-scale GBDP tree), average nucleotide identity (ANI) was calculated using FastANI.39 The reference genome sequence of S. kunmingensis DSM 41681 (ASM3561610v1) was obtained from publicly available genomic repositories, and the strain BTA 1-131 genome assembly was prepared using the following command: FastANI -q assembly_genome.fasta -r reference_genome.fasta -o ani_output.txt, where -q specifies the query genome (the polished genome assembly of the strain BTA 1-131), -r denotes the reference genome (S. kunmingensis DSM 41681), and -o defines the output file storing ANI results. The ANI percentage from the analysis result was used to determine the genomic relatedness between the assembly and the reference strain. Results were interpreted based on established ANI thresholds (90–95%) for bacterial species delineation.39
Sequencing of Streptomyces sp. BTA1-131 performed on both Illumina and ONT platform. The Illumina sequencing generated 11.25 million reads with a total base of 1.67 Gbp; the mean length was 149 bp, with a peak insert size of 261 bp. The filtering process resulted in 82.04% of reads (9.23 million reads) passing filtering (>Q30, >100 bp); the remaining reads were categorized as low quality (15.2%), too short (2.7%), and too many N (0.06%). The ONT sequencing generated 561,346 reads with a total base of 1.46 Gbp; reads N50 was 5,672 Kbp and more than 97% reads were >Q10. No filtering process was applied to ONT reads as the Flye assembler will use all data input from ONT raw reads.
The whole genome sequence of Streptomyces sp. BTA 1-131 was obtained from a total of 561,346 sequence reads from the ONT long-read assembly, followed by polishing with the Illumina short-read assembly. This hybrid assembly resulted in a 10,234,869 bp genome size with an N50 median of 9,496,435 bp and a GC content of 71.62%, consisting of one large chromosomal contig and three small contigs. Based on the CheckM analysis, the genome was 99.89% complete with a contamination level of 0.21%. In addition, the overall average coverage of the complete assembled genome was noted to be approximately 170.7X. This finding is supported by BUSCO result, 99.8% complete BUSCOs from Streptomyces_odb12 lineage dataset.
Moreover, both linear and circular topologies were found in the genome of Streptomyces sp. BTA 1-131. The BLAST analysis revealed that a linear chromosomal genome of the strain BTA 1-131 contains a long-terminal inverted repeat (L-TIR) of up to 1.5 Mbp at the chromosome end (9,496,435–7,999,105; 1,497,730 bp), resulting in a loop-like feature ( Figure 1). Two additional small linear contigs, 386.1 Kbp and 301.8 Kbp in size, along with the smallest circular contig at 50.5 Kbp, were identified as plasmids. In total, there were 8,986 protein-coding genes (CDSs) with the average open reading frame (ORF) length of 995 bp. Within the genome Streptomyces sp. BTA 1-131, 21 rRNA and 92 tRNA operons were also predicted ( Table 1).
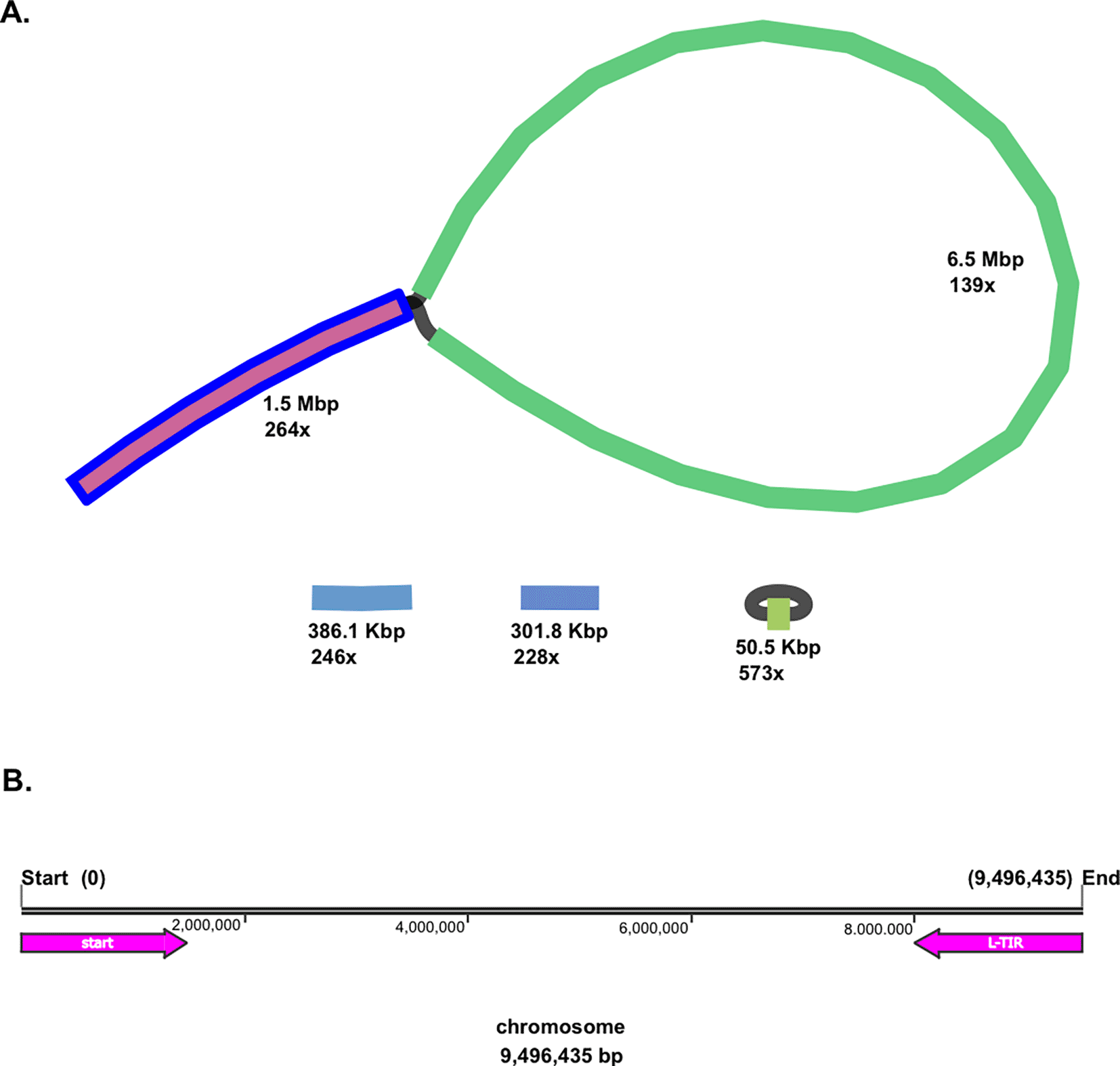
(A) Schematic representation of the Streptomyces sp. BTA 1-131 genome detected, adding up to 1.5 Mbp in length to chromosome size. In addition, a circular and two linear plasmids were detected from the strain BTA 1-131 assembly. (B) L-TIR is indicated with a reverse arrow (pink colour) from the end of chromosome (9,496,435–7,999,105; 1,497,324 bp).
| BTA 1-131 | DSM 41681 | NRRL B-1811T | |
|---|---|---|---|
| Genome topologya | linear | linear | linear |
| Contigs | 4 | 379 | 104 |
| Genome size (bp) | 10,234,869 | 9,797,351 | 7,633,340 |
| N50 | 9,496,435 | 57,076 | 245,715 |
| GC content (%) | 71.62 | 70.73 | 72.74 |
| Protein coding genes (CDS) | 8,986 | 8,633 | 6,405 |
| rRNA genes | 21 | 4 | 6 |
| tRNA genes | 92 | 83 | 81 |
| Genes assigned to COG | 87 | 78 | 36 |
| Label | Data | Type of data |
|---|---|---|
| Data file 1 | Figshare: Quality distribution of the BTA1-131 reads from Illumina and ONT sequencing This data contains the raw data read quality score, GC content (%), and general statistics of the reads https://doi.org/10.6084/m9.figshare.24204633 | Web report (HTML) Attribution: CC BY 4.0 |
| Data file 2 | Figshare: High-quality reads resulted from Illumina raw read filtering (Q > 20, length > 200) https://doi.org/10.6084/m9.figshare.24204720 | Reads (FASTQ) Attribution: CC BY 4.0 |
| Data file 3 | Figshare: Draft assembly from ONT raw read This data contains fasta file, graph file, and assembly statistic. https://doi.org/10.6084/m9.figshare.30563048 | Draft assembly (FASTA) Attribution: CC BY 4.0 |
| Data file 4 | Figshare: Topology hybrid assembly of Streptomyces sp. BTA 1-131 https://doi.org/10.6084/m9.figshare.30542942 | Image (PNG) Attribution: CC BY 4.0 |
| Data file 5 | Figshare: Hybrid assembly statistic report https://doi.org/10.6084/m9.figshare.24204789 | Report (ZIP) Attribution: CC BY 4.0 |
| Data file 6 | Figshare: Identification of BUSCO genes to assess genome completeness https://doi.org/10.6084/m9.figshare.24204855 | Report (TXT) Attribution: CC BY 4.0 |
| Data file 7 | Figshare: Genome completeness and contamination assessment with CheckM https://doi.org/10.6084/m9.figshare.30563456 | Report (TXT) Attribution: CC BY 4.0 |
| Data file 8 | Figshare: Genome visualisations including identified COGs https://doi.org/10.6084/m9.figshare.30564893 | Report (PDF) Attribution: CC BY 4.0 |
| Data file 9 | Figshare: Identification for potential new species using TYGS https://doi.org/10.6084/m9.figshare.30563564 | Report (PDF) Attribution: CC BY 4.0 |
| Data set 1 | NCBI GenBank: The nucleotide sequence of 16S rRNA gene of the strain BTA 1-131 NCBI accession number MT280129.1 https://www.ncbi.nlm.nih.gov/nuccore/MT280129 | Contigs (FASTA) |
| Data set 2 | The cultivable isolate: The Indonesian Culture Collection BRIN (InaCC), accession number A1205 This data contains a deposition data sheet of the isolate BTA 1-131 in InaCC https://hdl.handle.net/20.500.12690/RIN/UKGE4I | Report (PDF) Attribution: CC BY-NC-ND 4.0 |
| Data set 3 | Raw reads from Illumina and ONT sequencers This data contains raw reads deposited in the DNA Data Bank of Japan (DDBJ) and Indonesian national scientific repository (RIN Dataverse) DDBJ Bioproject: PRJDB16533 https://ddbj.nig.ac.jp/search/entry/bioproject/PRJDB16533 RIN Dataverse: https://hdl.handle.net/20.500.12690/RIN/UQP1WS | GenBank collection (FASTQ) |
Furthermore, the general genome features of Streptomyces sp. BTA 1-131 are summarised in comparison to its closely related neighbour strain S. kunmingensis DSM 41681 and the type strain S. albus NRRL B-1811 in Table 1, while the schematic of circular maps of the genome, including the annotated COG categories, is presented in Figure 2.
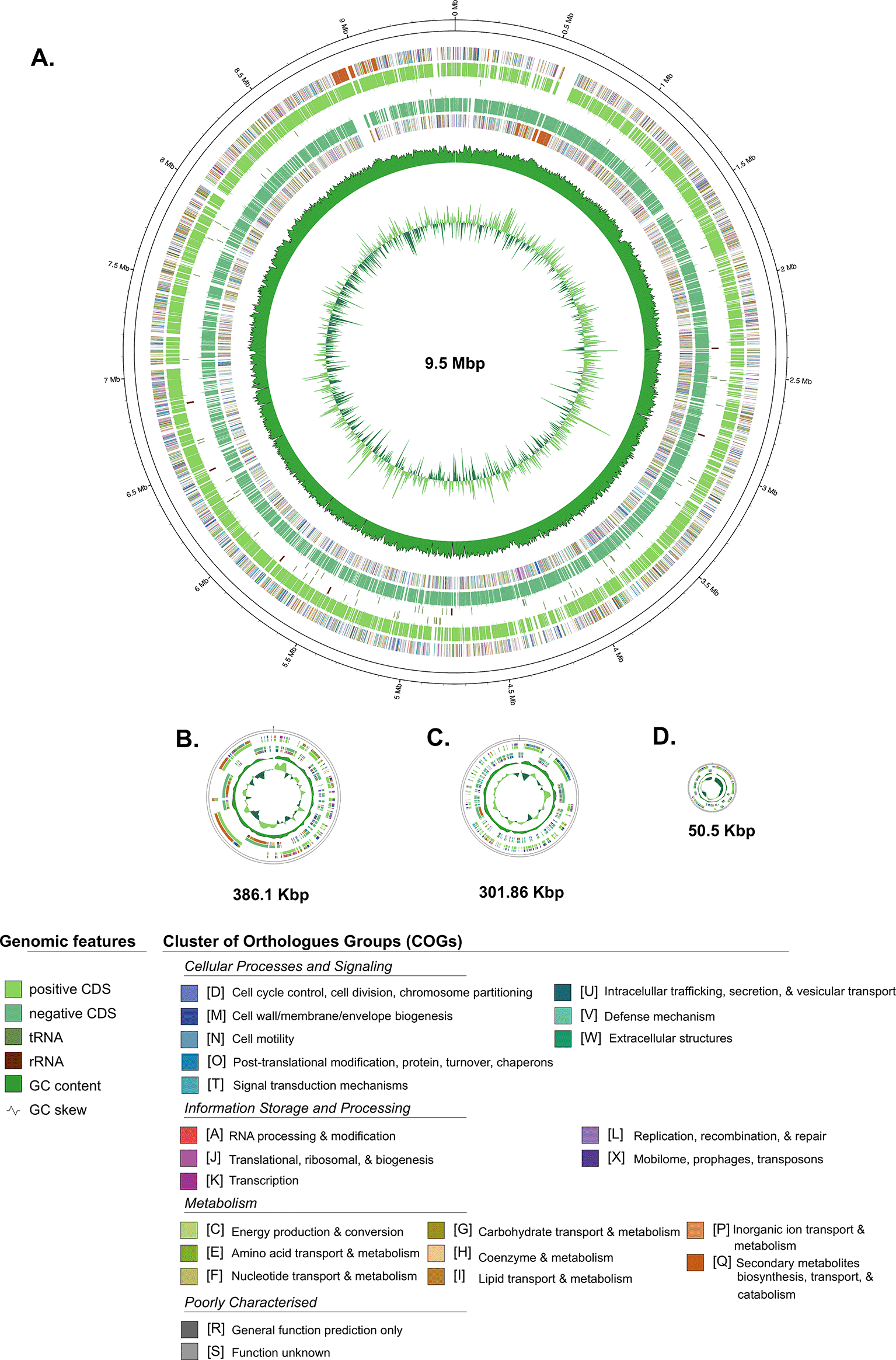
Contig of L-TIR displayed together as one chromosome. Labelling from outside to the inside: contigs; COGs on the forward strand; CDS, rRNAs, and tRNAs on the forward strand; CDS, rRNAs, and tRNAs on the reverse strand; GC content; GC skew.
As aforementioned, the strain BTA 1-131 was previously identified by sequencing the 16S rRNA gene and is closely related to S. kunmingensis NBRC 14463 with the sequence similarity of 98.4%. Indeed, the results of TYGS species identification suggested a potential novel Streptomyces species for the strain BTA 1-131, as indicated by the dDDH value below the threshold for classification as the same species as the nearest strain, S. kunmingensis DSM 41681 (<70%) ( Figure 3). Moreover, the ANI score between the BTA 1-131 genome assembly and S. kunmingensis DSM 41681 was 89.15%.
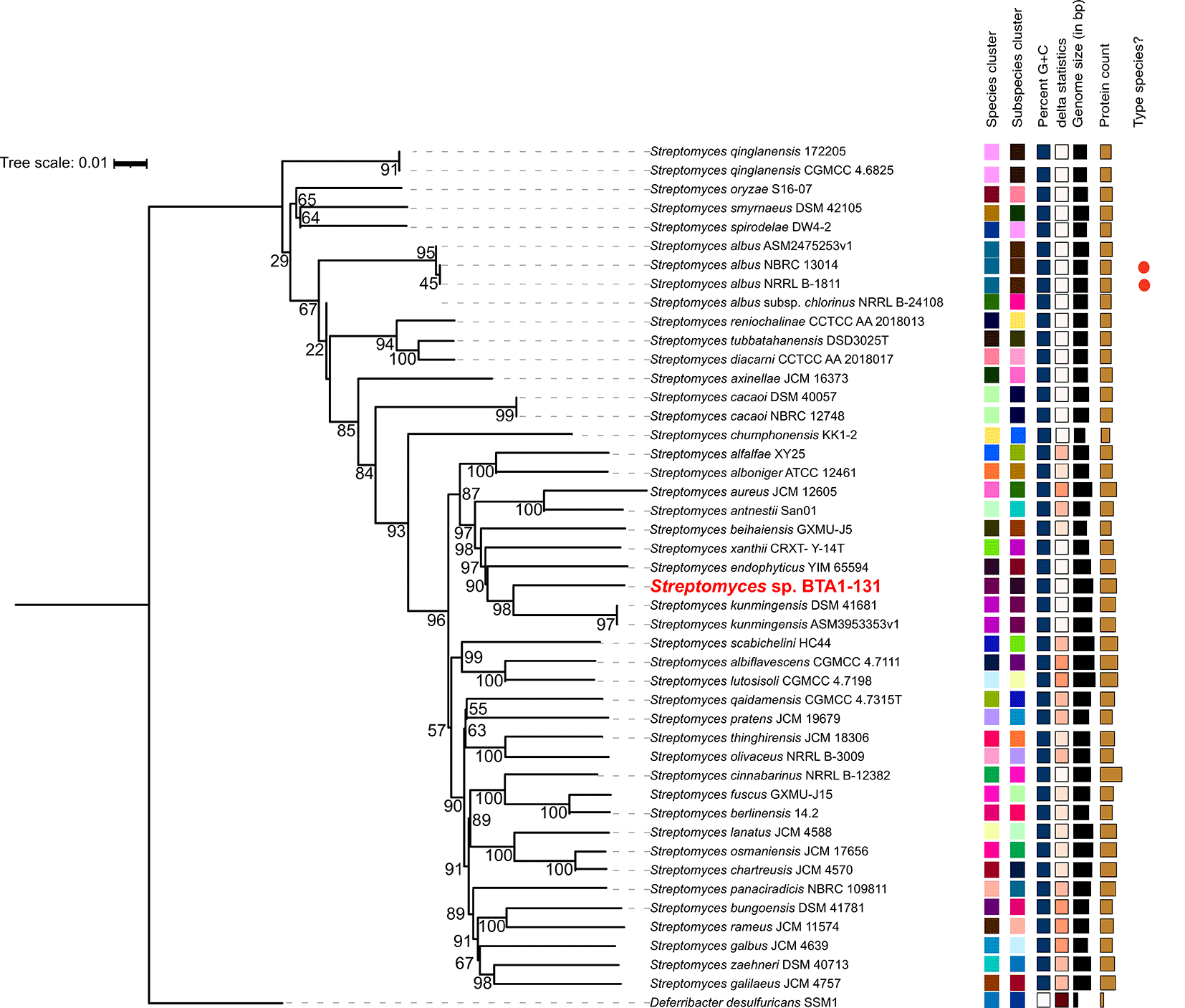
The main limitation in this study is that although the phylogenomic analysis showed a possibility of the strain BTA 1-131 as a new Streptomyces species, a follow-up taxonomical confirmation through polyphasic study is necessary to establish its species status. Additional taxonomical investigations, including chemotaxonomic, morphological, and biochemical characterisation, would be required to definitively confirm the novelty of this strain and validate its classification within the genus Streptomyces.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)