Keywords
CB1 agonists, pharmacophore based virtual screening, lead discovery, quinazoline-2,4(1H,3H)-dione derivatives
This article is included in the Cheminformatics gateway.
CB1 agonists, pharmacophore based virtual screening, lead discovery, quinazoline-2,4(1H,3H)-dione derivatives
Details of the manuscript update
Introduction section:
In the paragraph after Figure 1, line 2: "The AM11542CB1" was changed to "The AM11542-CB1".
In the paragraph after Figure 1, line 7: " is most likely do to the flexibility " was changed to " is most likely due to the flexibility ".
In Table 2 : "Kcal" was changed to "kcal" to correct the unit notation.
See the authors' detailed response to the review by Samir Chtita and Bouchra Rossafi
See the authors' detailed response to the review by Mourad Fawzi
See the authors' detailed response to the review by Fatima En-nahli
Cannabinoid receptors are classified as G protein-coupled receptors which belong to a family known as the endocannabinoid system, a master regulator of numerous physiological pathways throughout the human body.1 There are two categories of cannabinoid receptors, known as CB1 and CB2. The CB1 receptor is found throughout multiple organs of the body; it is present in the digestive tract, liver, pancreas, and musculature, in addition to its primary site of localization being in the brain.2 In fact, CB1 is the most highly expressed receptor in the brain relative to other receptors examined, possessing 7 transmembrane domains with G protein coupling. The CB1 receptor mediates cannabinoid-induced psychotropic effects. The CB2 receptor is more so located within immune cells, where it exerts its immunomodulatory role.
Cannabinoids are a class of chemical compounds which are championed globally for their psychoactive and physiologic whole body functions; for at least 5,000 years,3,4 mankind has been able to capitalize on the value of cannabinoids. One such natural source of cannabinoids is cannabis, known as “kif” in Moroccan culture, a natural product with a plethora of phytoconstituents including Δ9-tetrahydrocannabinol (THC). The phytochemical THC exerts its psychotropic effects by acting at the CB1 receptor, which also serves as the primary receptor for the endogenous endocannabinoids anandamide (AEA) and 2 arachidonoylglycerol (2-AG).5 Activation of the CB1 receptor upregulates potassium channel currents and calcium ion channel polarization, making receptor signaling dose dependent and responsive to treatment with pertussis toxin.6 In addition, CB1 can exist as a homodimer and/or form complexes as heterodimers or heterooligomers with other G Protein-Coupled Receptors (GPCRs). Lastly, the CB1 receptor is in a complex with GABAergic and glutamatergic cells, and therefore, CB1 receptor stimulation decreases release from GABAergic and glutamatergic cells.7
The structural complexes of the cannabinoid receptor CB1 with THC analogs are an important topic of research to help us understand the molecular interactions that underlie cannabinoid signaling. Thus far, the repertoire of known complexes of the CB1 receptor includes the structure of CB1, bound to AM8411,8 CP559409 and AM1154210 ( Figure 1). These structures have a resolution of 2.8-3.4 Å and provide a highly informative account of the binding modes and associated conformational changes that can accompany receptor activation. Several important interacting residues have been found to be conserved between agonist-bound structures, including hydrophobic interactions with LEU1933.29, VAL1963.32, PHE2003.36, TYR2755.39, LEU2765.40, TRP2795.43, TRP3566.48, LEU3596.51 and MET3636.55.11 These observations further emphasize the importance of these residues in their role of stabilizing binding of agonists to the receptor, while also promoting receptor activation.
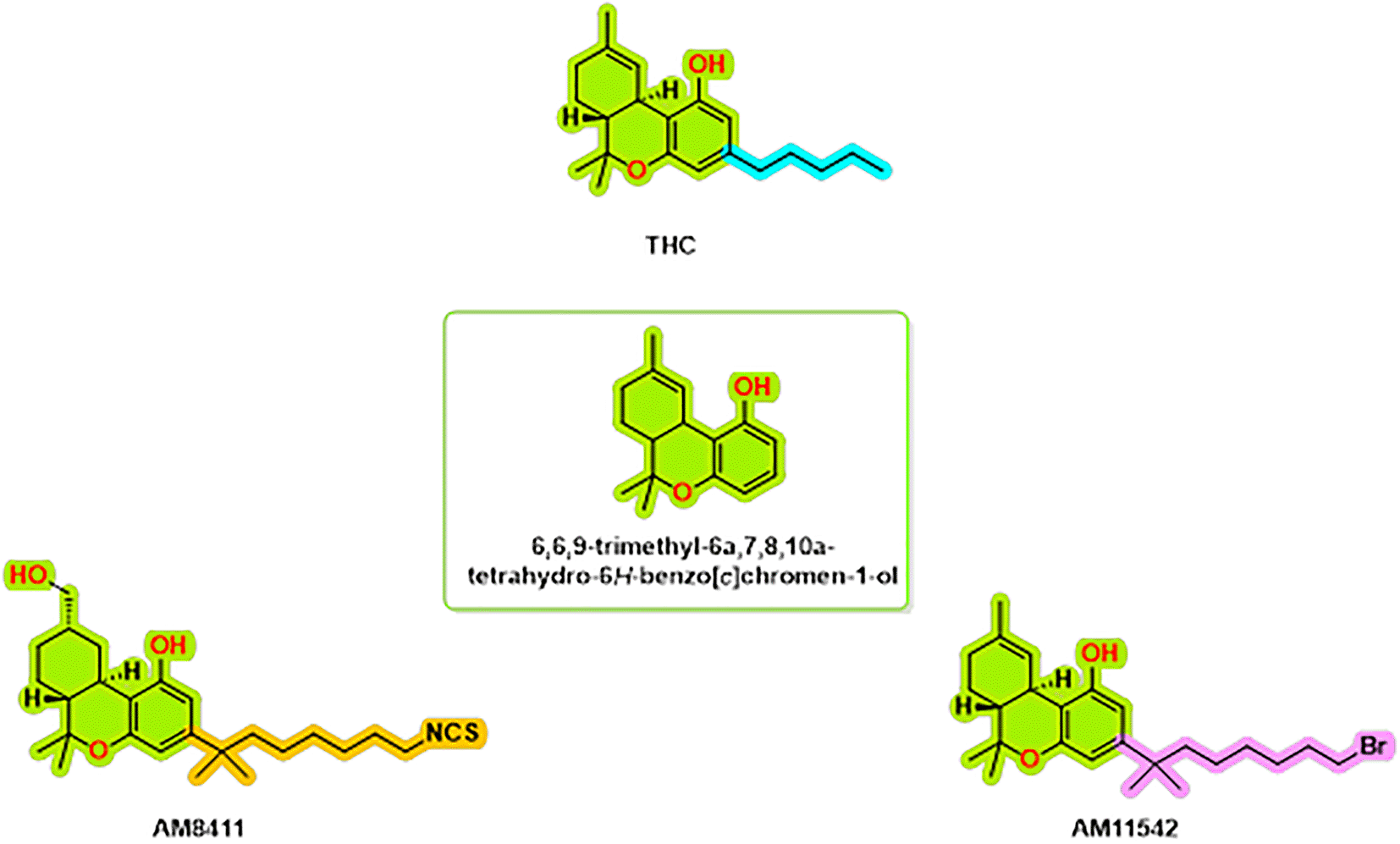
There were also three structures solved by means of cryo-electron microscopy (cryo-EM) that included full active states along with the Gi1-2 G protein subunit.12 The AM11542-CB1 complex is a crystalline structure and had a fusion protein, flavodoxin, which contributes to stabilization of TM6 in an active confirmation.11 All of the structural coordinates cover the full CB1 protein sequence, covering anywhere between approximately 58% and 62% of each residues in all active structures.8 The fact that the coordinates do not cover the full sequence is most likely due to the flexibility imparted by both the N and C-terminus and the long three intracellular loops (ICL3). These structural descriptions not only allow constructs for determining the molecular mechanisms for THC analog activation of the CB1 receptor but also create opportunities for future studies.
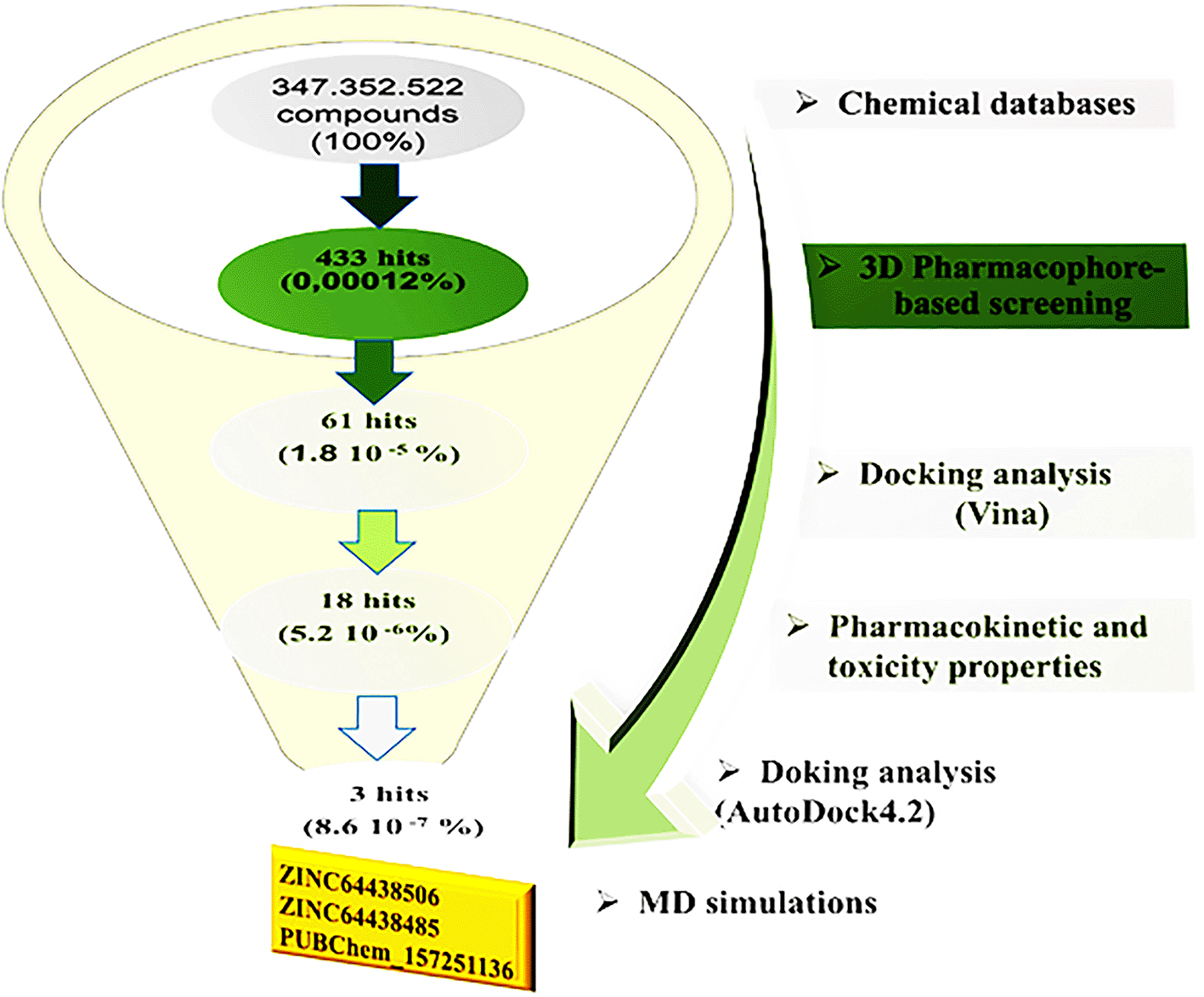
In recent years the harmonization of approaches have created several avenues to create CB1 receptor agonists with less adverse effects. This study will previously develop a CB1 agonist pharmacophore model that was used as a virtual screening tool for unique/novel class of CB1 agonists. Using the pharmacophore this was able to screen over 300 million hits, of compounds, parsed from 11 different databases and were able to identify many candidates that have not been screen for cannabinoid receptor binding assay. This highlights the usefulness of computational strategies to find novel therapeutics that have the potential to provide safer alternatives.
Pharmacophore based virtual screening were performed using the Pharmit web interface (http://pharmit.csb.pitt.edu/), which has a number of built-in databases that provide access to comprehensive data; Molprot (4,742,020 molecules), ChEMBL34 (2,264,112 molecules), ZINC (13,127,550 molecules), ChemDiv (1,456,120 molecules), ChemSpace (50,181,678 molecules), Enamine (4,117,328 molecules), MCULE (39,843,637 molecule), MCULE-ULTIMATE (126,471,502 molecules), NCI Open Chemical Repository (52,237 molecules), LabNetwork (1,794,286 molecules), and PubChem (103,302,052 molecules). The pharmacophore model was constructed using selected PDB code 5XRA from the RCSB Protein Data Bank (http://www.rcsb.org/structure/5xra), with agonist AM11542. The model utilized a pharmacophore framework built on five features, adhering to the default parameters of the Pharmit server. The Pharmit filters were applied based on the Lipinski rule of five and Veber’s rule to refine the screening process and identify the most selective CB1 agonist. Figure 2 illustrates the multi-step virtual screening process used in this work.
The ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) filter is one of the phases of the drug discovery and development process.13–15 This allows researchers to determine potentially druggable drug-like properties and toxicology early on to assess which compounds will be effective drugs with safety potential. Thus, relative to virtual screening, such a filter can detect adverse properties that would fail at much later stages, such as insufficient absorption, excessive toxicity, and/or suboptimal bioavailability. Two of the trusted websites relied upon to evaluate such properties based upon lipophilicity, logP, solubility, etc., are SwissADME (http://www.swissadme.ch) and pKCSM (https://biosig.lab.uq.edu.au/pkcsm/prediction). Therefore, when these are filtered out early on, time and costs will be effectively saved down the line.
To estimate the binding affinities of the 433 highest-ranked compounds to CB1, molecular docking was carried out using AutoDock Vina integrated within the PyRx platform. The crystal structure of CB1 (PDB ID: 5XRA), with a resolution of 2.8 Å,8,16,17 was retrieved from the Protein Data Bank. Before docking, water molecules and the native ligand were removed from the protein, followed by the addition of polar hydrogen atoms and Kollman charges. The prepared protein structure was then energy-minimized using UCSF Chimera and saved in PDBQT format.
Prior to docking, all 433 compounds underwent pre-optimization utilizing the Universal Force Field (UFF) in combination with the conjugate gradient algorithm.18 Optimization parameters were set to 2000 total steps, with updates occurring every step, and the process was programmed to terminate once the energy difference fell below 0.01 kcal/mol.19 After optimization, the compounds were converted to PDBQT format and docked at specific binding site coordinates (x = -42.052, y = -164.338, z = 306.631). Molecules demonstrating the lowest binding energies and root-mean-square deviation (RMSD) values below 2 Å were selected for subsequent analyses.
Molecular dynamics (MD) simulations were carried out on the top-ranked docking conformations using the GROMACS 2024.4 software package, employing the CHARMM27 all-atom force field.20 The topology files for the CB1 receptor were generated using the pdb2gmx tool,21 while the ligand topologies were prepared through the CHARMM General Force Field (CGenFF) using the Param-Chem server.22–24 Each receptor-ligand complex was embedded in a triclinic simulation box and solvated with TIP3P water molecules, maintaining a minimum distance of 1.0 nm from the box boundaries. To neutralize the systems, Na+ and Cl- ions were added accordingly.
Prior to the production runs, energy minimization was performed using the steepest descent algorithm for 50,000 steps to eliminate steric clashes and ensure system stability. Subsequently, two equilibration phases were conducted: first under the NVT ensemble to stabilize temperature, followed by the NPT ensemble to equilibrate pressure and density. Input files for both equilibration and production stages were customized to adjust parameters such as trajectory-saving intervals, energy monitoring, and other essential simulation settings. Finally, each ligand-receptor system underwent 100 ns of production MD simulation at a constant temperature of 300 K, pressure of 1 bar, and a time step of 2 femtoseconds.
The binding free energy was calculated via MM/GBSA (Molecular Mechanics Generalized Born Surface Area).25 This approach utilizes force fields of molecular mechanics and an implicit solvent approach to conclude stability and binding tendencies of molecular complexes. It is particularly useful for classifying ligands and understanding overall energetic contributions of molecular recognition. The binding free energy ΔGbind of a complex can be calculated as follows:
Where TΔS, ΔEMM and ΔGsol are the conformational entropy upon binding, the changes of the gas phase molecular mechanism (MM) energy and the solvation free energy, respectively.
The pharmacophore model of the CB1 receptor agonist was generated based on the AM11542 agonist. The results are shown in Figure 3. A five-point model with 1 aromatic, 2 hydrogen bond acceptors and 2 hydrophobic regions was generated using the Pharmit web server.26
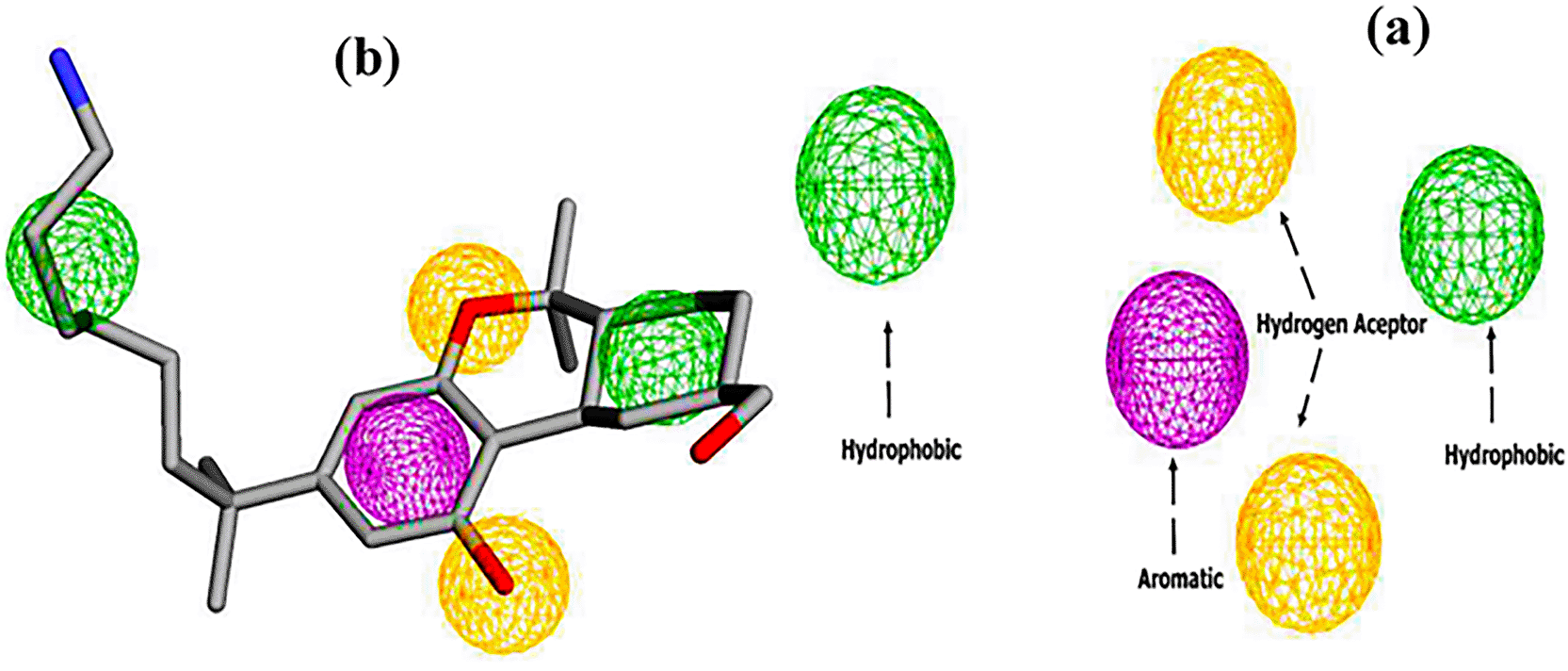
Subsequently, virtual screening was performed across 11 databases, identifying 433 compounds containing molecular groups that matched the pharmacophore mode.
Pharmacophore-based virtual screening was performed using information from the previous pharmacophore model. Approximately 300 million compounds from the Pharmit database were filtered. The criteria for filtering the library were as follows: molecular weight limits were set at 250-500 Da, the maximum number of hydrogen bond donors was less than 4, the maximum number of hydrogen bond acceptors was less than 9, the maximum number of rotatable bonds was less than 9, Log P was between 2 and 5, and polar surface area was less than 140 Å. The results are computed and classified according to different criteria such as energy minimization. The 433 top-ranked compounds are presented in Table 1.
Next, molecular docking analysis was performed using AutoDock Vina, implemented in the PyRx software. Compounds with a binding affinity of less than - 9.00 kcal/mol were selected for further analysis, while the others were excluded. Additionally, the predicted binding modes were required to have a root-mean-square deviation (RMSD) of less than 2Å when superimposed onto the native agonist (AM11542). The top 61 selected compounds are listed in Table 2, while the remaining candidates are provided in Table S1 (see supplementary data).
The ranked compounds from docking analysis were evaluated for potential toxicity, including an AMES toxicity test, an acute oral toxicity test in rats (LD50), a skin sensitization test and a maximum tolerated dose analysis. Highly toxic compounds are not considered in further studies. The top selected compounds are presented in Table 3. The other are presented in the supplementary data ( Table S2).
AMES toxicity, oral rat acute toxicity (LD50), and Max Tolerated dose in humans.
As described in Table 3, all selected compounds exhibited no Skin Sensitisation and no AMES toxicity. Additionally, the LD50 values for oral rat toxicity, ranging from 1.439 to 3.354 mol/kg, suggest moderate acute toxicity levels, consistent with safety margins suitable for therapeutic use. Moreover, the maximum tolerated dose in humans’ ranges from -1.12 to 1.059 Log mg/kg/day.
Physicochemical properties were evaluated using Lipinski’s rule of five,27 Ghose’s rule,28 Veber’s rule,29 Egan’s rule30 and Muegge’s rule,31 with results detailed in Table 4. Based on these guidelines, it is suggested that for a compound to be effectively absorbed and administered orally, it must meet specific physicochemical parameters. These criteria serve as essential benchmarks for assessing the compound’s potential for bioavailability and oral absorption. Compounds with two or more violations are not considered in the further analysis (see Table S3 in supplementary data).
To predict the pharmacokinetic properties of absorption, distribution, metabolism and excretion (ADME), pkCSM web server was used to calculate the following parameters: water solubility (log mol/L), Caco-2 cell permeability, human intestinal absorption (HIA), blood-brain barrier (BBB) permeability, central nervous system (CNS) permeability and total clearance.
Water solubility (LogS) indicates the solubility of a compound in water at 25°C. Generally, water-soluble drugs are more readily absorbed than lipid-soluble ones. in vitro Caco-2 cell permeability is a crucial measure of drug absorption, with a compound considered to have high Caco-2 permeability if its value surpasses 8 x 10−6 cm/s. Within the pkCSM model, high Caco-2 permeability aligns with predicted values exceeding 0.90. The intestine typically serves as the primary site for drug absorption from orally administered solutions. A compound with absorbance below 30% is deemed poorly absorbed. An established gauge of blood-brain barrier (BBB) penetration is the log BB ratio, which reflects drug molecule concentrations in the brain and blood. Compounds with log BB > 0.3 exhibit high BBB permeability, while those with log BB < 1.0 show limited BBB distribution. Furthermore, central nervous system (CNS) permeability is a vital parameter for assessing the blood-brain permeability of a drug candidate, expressed as LogPS. Compounds with LogPS <-3 are considered incapable of penetrating the CNS.
Based on these ADME properties, 18 compounds were selected for further analysis. The results for these compounds are detailed in Table 5, while the other parameters are shown in Table S4 in the supplementary data.
Docking validation was performed using AutoDock 4.2. The results are presented in Table 6.
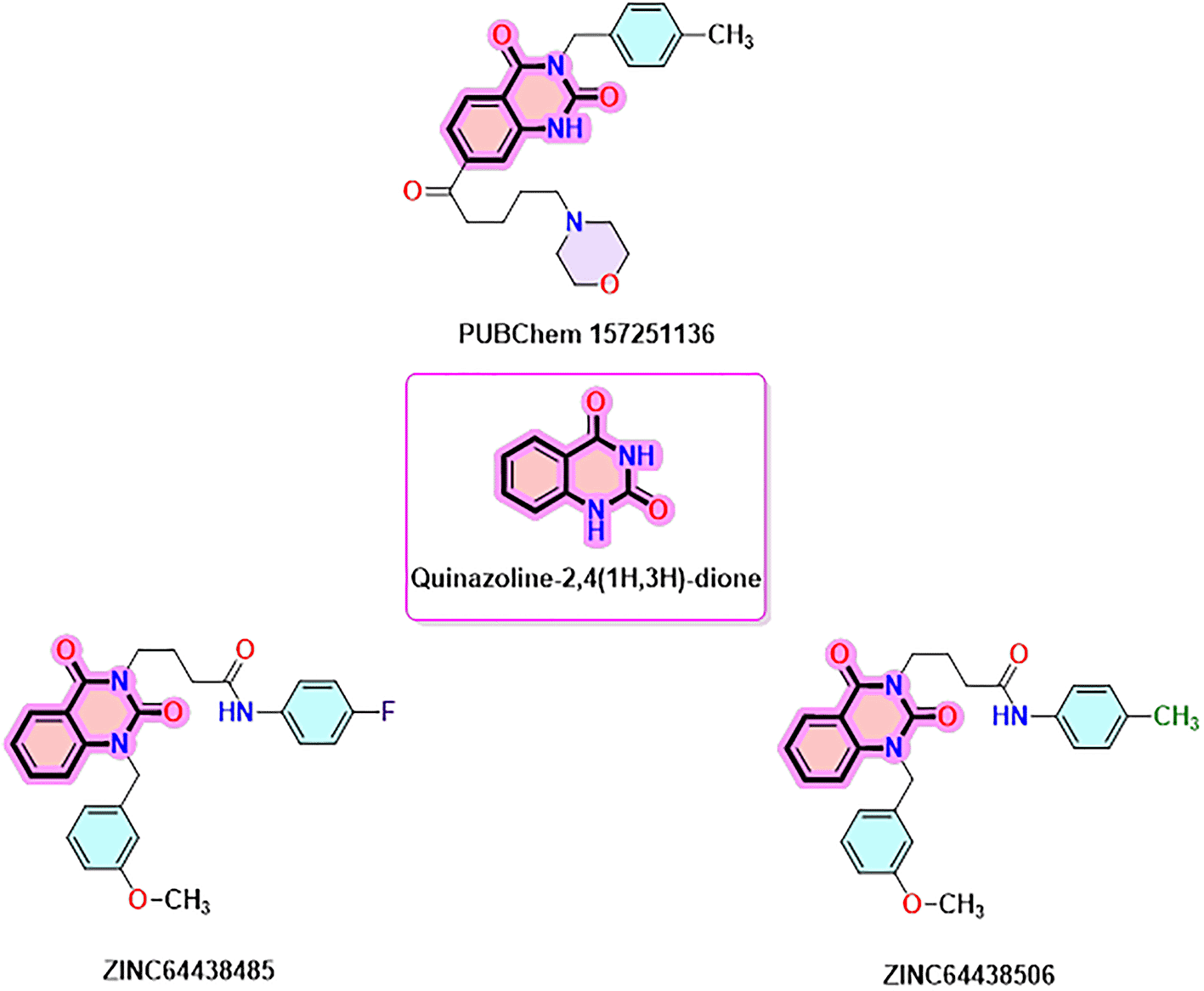
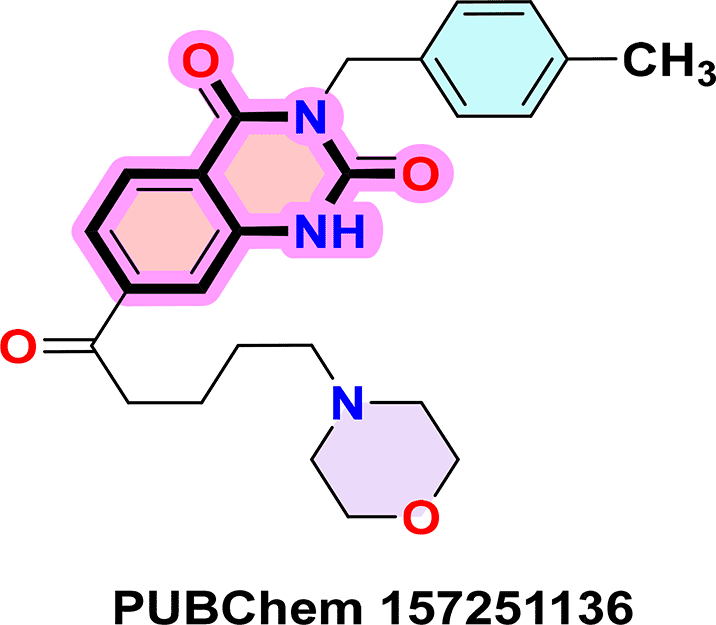
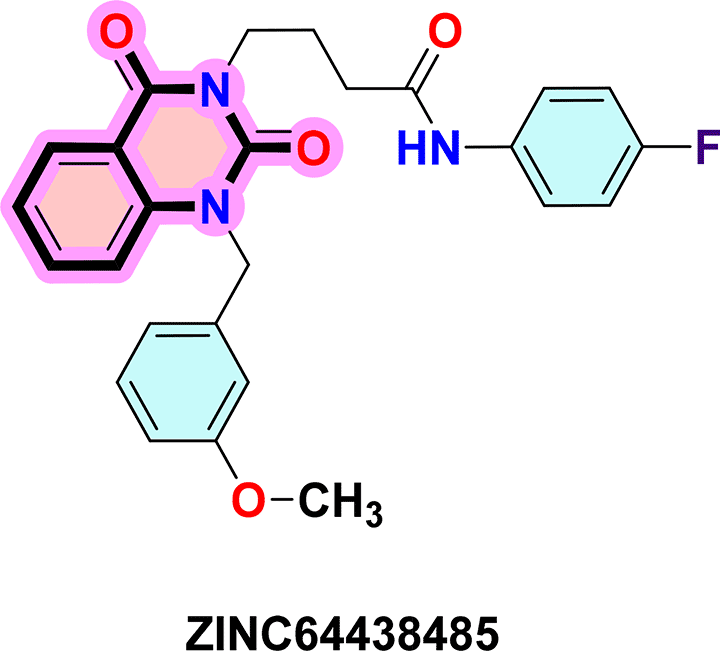
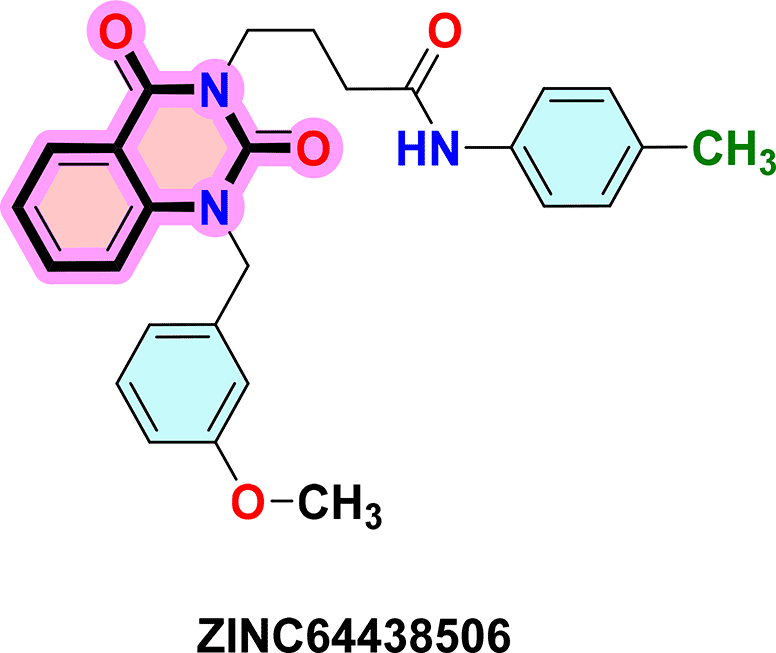
To identify the top CB1 agonist several criteria were taken into account including energy values (e.g. binding energy or Ki values), interactions with key amino acids in the CB1 binding site, number of hydrogen bonds and distances between hydrogen bonds. These interactions are essential for designing effective and selective CB1 agonists.10 For instance, π-π interactions with aromatic residues (Phe296, Phe170, Phe174, Trp 279) and hydrogen bonds with Ser383 or Thr197 stabilize the agonist-receptor complex, while residues such as Phe379 and Asp366 play a key role in receptor activation through electrostatic interactions.10 Based on these criteria three compounds were identified as CB1 receptor agonists: ZINC64438506, ZINC64438485 and PUBChem157251136.
ZINC64438506: The docking analysis of CB1 receptor and ZINC64438506 selected agonist is shown in Table 7 and Figure 4. The ZINC64438506 agonist was fixed in the CB1 binding pocket (cavity size = 2963 Å) through various type of interactions, including hydrogen bonds with key amino-acid residues SER A: 173 and SER A: 383, hydrophobic interactions with residues PHE A: 108, PHE A: 177, PHE A: 189, LEU A: 193, THR A: 197, PHE A: 268, TYR A: 275, LEU A: 276, TRP A: 279 and PHE A: 379 and π-π interaction with PHE A: 268. These interactions likely contribute to the compound’s low binding affinity (-13.11 Kcal/mol) and low inhibition constant (Ki = 0.244 nM) ( Table 6). The strong binding affinity may be attributed to the presence of short hydrogen bonds with SER A: 173 (3.34 Å) and SER A: 383 (51.79 Å).
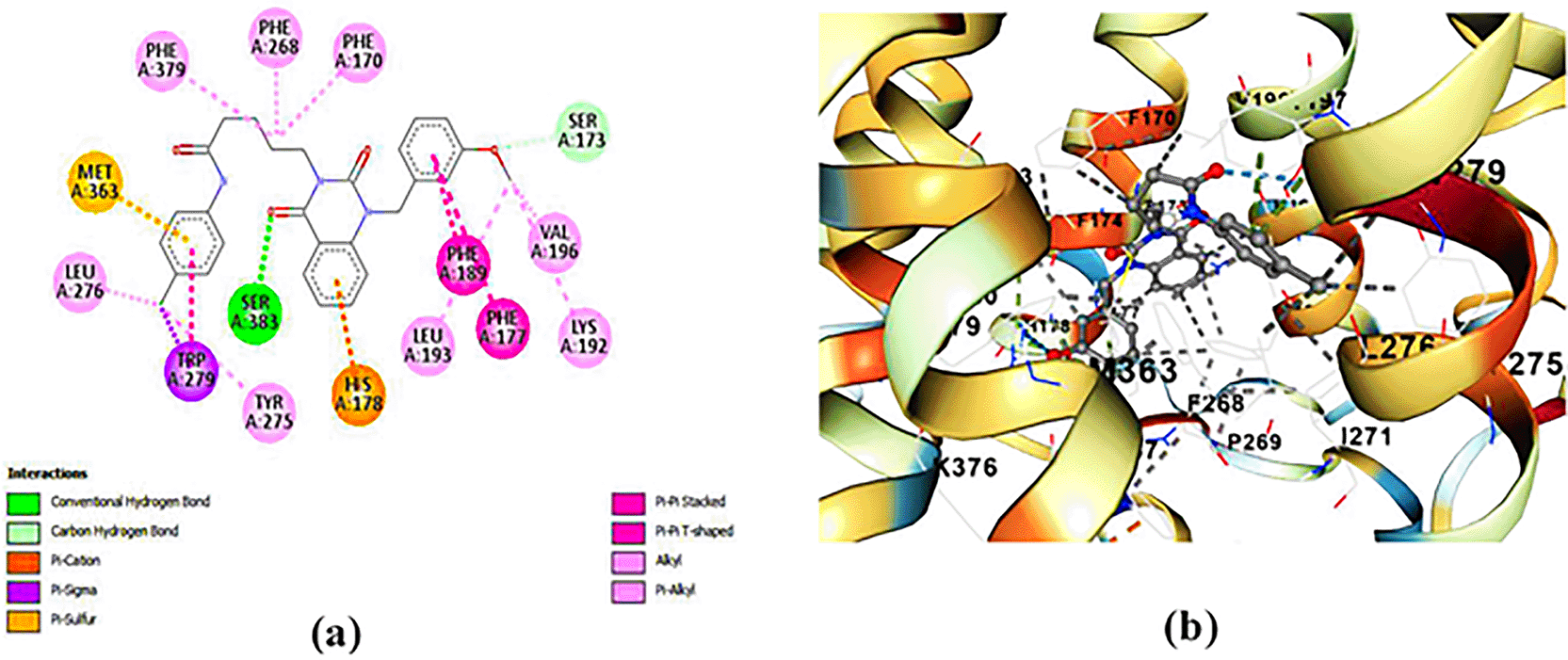
(a) 2D view of binding site interactions (b) 3D view of binding conformation.
ZINC64438485: The docking analysis of CB1 receptor and ZINC64438485 selected agonist is shown in Table 7 and Figure 5. The ZINC64438485 agonist was fixed in the CB1 binding pocket (cavity size = 2963 Å) through various type of interactions, including hydrogen bonds with key amino-acid residues THR A: 197 and SER A: 383, hydrophobic interactions with residues PHE A: 174, PHE A: 177, PHE A: 189, LEU A: 193, VAL A: 196 and ILE A: 271 and π-π interaction with PHE A: 174, HIS A: 178, PHE A: 268 and TRP A: 279. These interactions likely contribute to the compound’s low binding affinity (-13.07 Kcal/mol) and low inhibition constant (Ki = 0.262 nM) ( Table 6). The strong binding affinity may be attributed to the presence of short hydrogen bonds with THR A: 197 (1.94 Å) and SER A: 383 (3.08 Å).
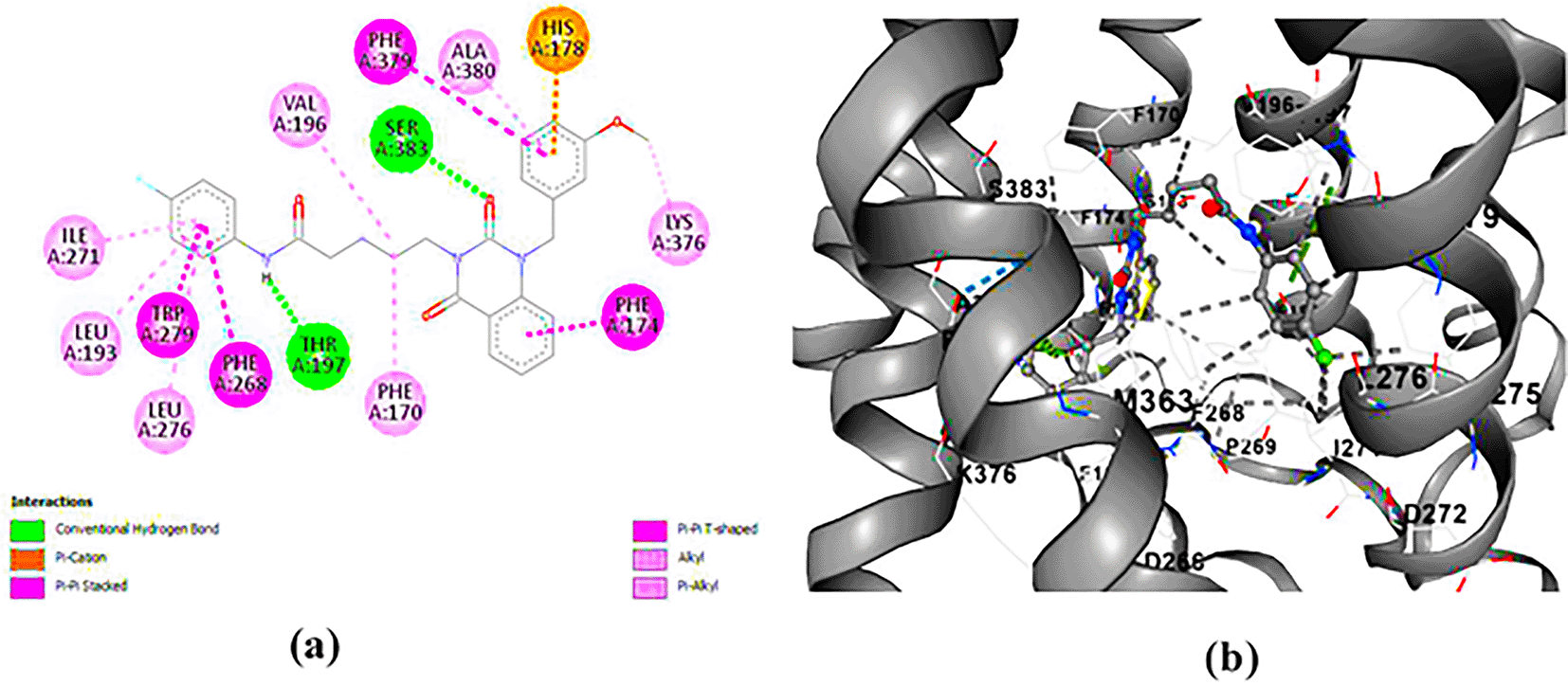
(a) 2D view of binding site interactions (b) 3D view of binding conformation.
PUBChem-157251136: The docking analysis of CB1 receptor and PUBChem-157251136 selected agonist is shown in Table 7 and Figure 6. The PUBChem157251136 agonist was fixed in the CB1 binding pocket (cavity size = 2963 Å) through various type of interactions, including hydrogen bonds with key amino-acid residues SER A: 173, HIS A: 178 and SER A: 383, hydrophobic interactions with residues PHE A: 174, PHE A: 177, LEU A: 193, THR A: 197, PHE A: 268, LEU A: 276, TRP A: 279 and PHE A: 379 and π-π interaction with PHE A: 268 and TRP A: 279. These molecular interactions likely contribute to the compound’s low binding affinity (-11.84 Kcal/mol) and low inhibition constant (Ki = 2.09 nM) ( Table 6). The strong binding affinity may be attributed to the presence of three hydrogen bonds with SER A: 173 (3.23 Å), HIS A: 178 (1.94 Å) and SER A: 383 (2.04Å).
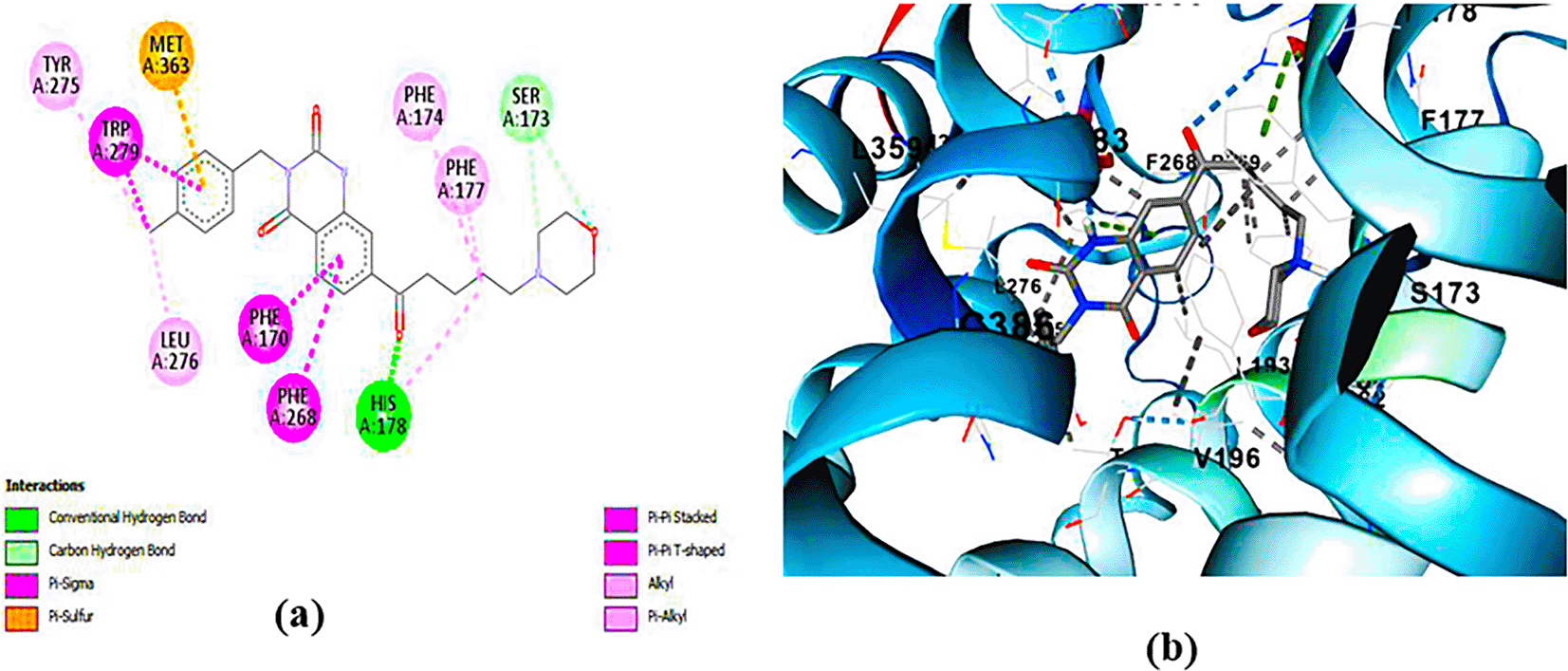
(a) 2D view of binding site interactions (b) 3D view of binding conformation.
While molecular docking analyses are enough to understand possible bindings based on ligand positioning and receptor-ligand interactions, it should be noted that such methodologies evaluate the flexibility of the ligand only while keeping the protein in a rigid form. Thus, in order to evaluate the best-docked candidates for binding pose stability and dynamics of protein conformation, molecular dynamics (MD) simulations were performed over 100 ns. The results of the MD simulations, including root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and Ligand-Receptor Interaction Plot analyses, can be found in Figures 7–10. These studies provide insights into the dynamic behavior and stability of the protein–ligand complexes over time.
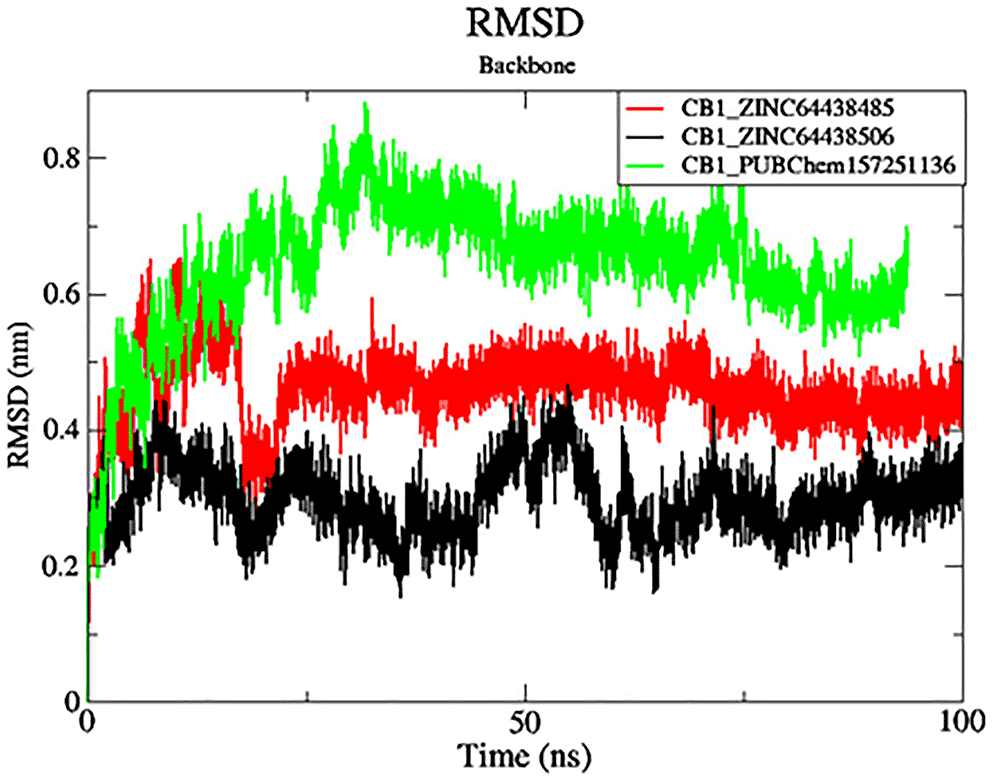
The black color was the CB1_ZINC64438506 complex, the red was the CB1_ZINC64438485 complex and the green was the CB1_PUBChem157251136 complex.
7.1 Root Mean Square Deviation (RMSD)
RMSD analysis was carried out for the protein backbone to have an idea of each protein-ligand complex’s structural stability during the simulation.32,33 The results are shown in Figure 7. As shown in Figure 7, mean RMSD values for the CB1_ZINC64438506, CB1_ZINC64438485 and CB1_PUBChem157251136 complex are 0.29 nm, 0.45 nm and 0.64 nm, respectively. The global RMSD values for CB1_ZINC64438506, CB1_ZINC64438485 are small which indicates that these ligands remain stable during the simulation of 100 ns and remained in the binding pocket of the CB1 receptor. The stability of the ligands can be attributed to several strong hydrogen bonds being mediated between these agonists and some of the key amino acids positioned within the binding pocket of the CB1 receptor.
7.2 Root mean square fluctuation (RMSF)
Root mean square fluctuation (RMSF) was used to determine the rigid and flexible regions of the CB1 receptor over the 100 ns of MD simulations.14 RMSF has been term definition, instead of just RMSD values, so that the maximum range of motion of a bound ligand can be seen. RMSF is defined as a standard measure of deviation of a molecule from its initial position.34 Molecules and residues should not present a high value, which indicates a flexibility, and those that appears low value has a greater rigidity. The RMSF plot for all complexes ( Figure 8) indicates that most residues located in the CB1 receptor has a low RMSF value, indicating they were rigid and retained stability over the entire 100 ns MD simulation.
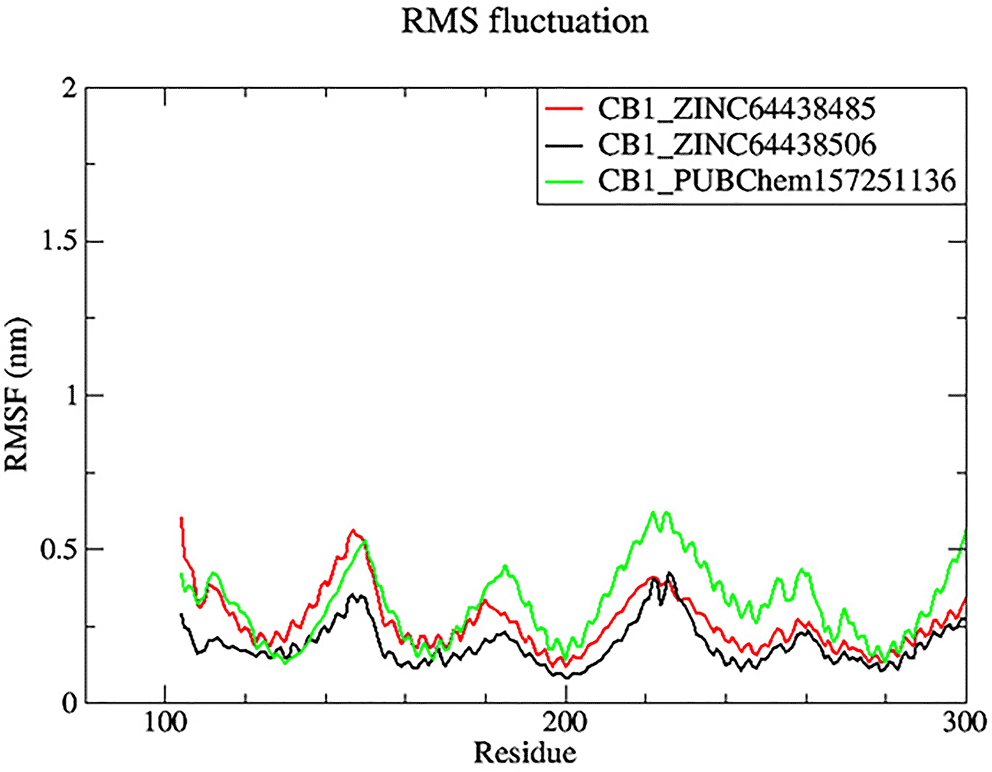
The black color was the CB1_ZINC64438506 complex, the red was the CB1_ZINC64438485 complex and the green was the CB1_PUBChem157251136 complex.
7.3 Radius of gyration (Rg)
The radius of gyration (Rg) is another important metric measured during MD simulations to determine spatial characteristics of a protein-ligand complex. The radius of gyration is the root mean square distance of all atoms making up a given structure from a relative center common point (the center of mass) and signifies extension versus folding of the given structure. Therefore, a lower Rg would imply stability and a more tightly folded structure, while a higher Rg would imply extension and flexibility of possible structures. In this work, the Rg profiles remained stable over the course of 100 ns of MD simulation for the complexes CB1_ZINC64438506 (mean = 4.70 nm; Figure 9) and CB1_ZINC64438485 (mean = 4.72 nm), while the complex CB1_PUBChem157251136 decreased in Rg (mean = 2.81 nm).
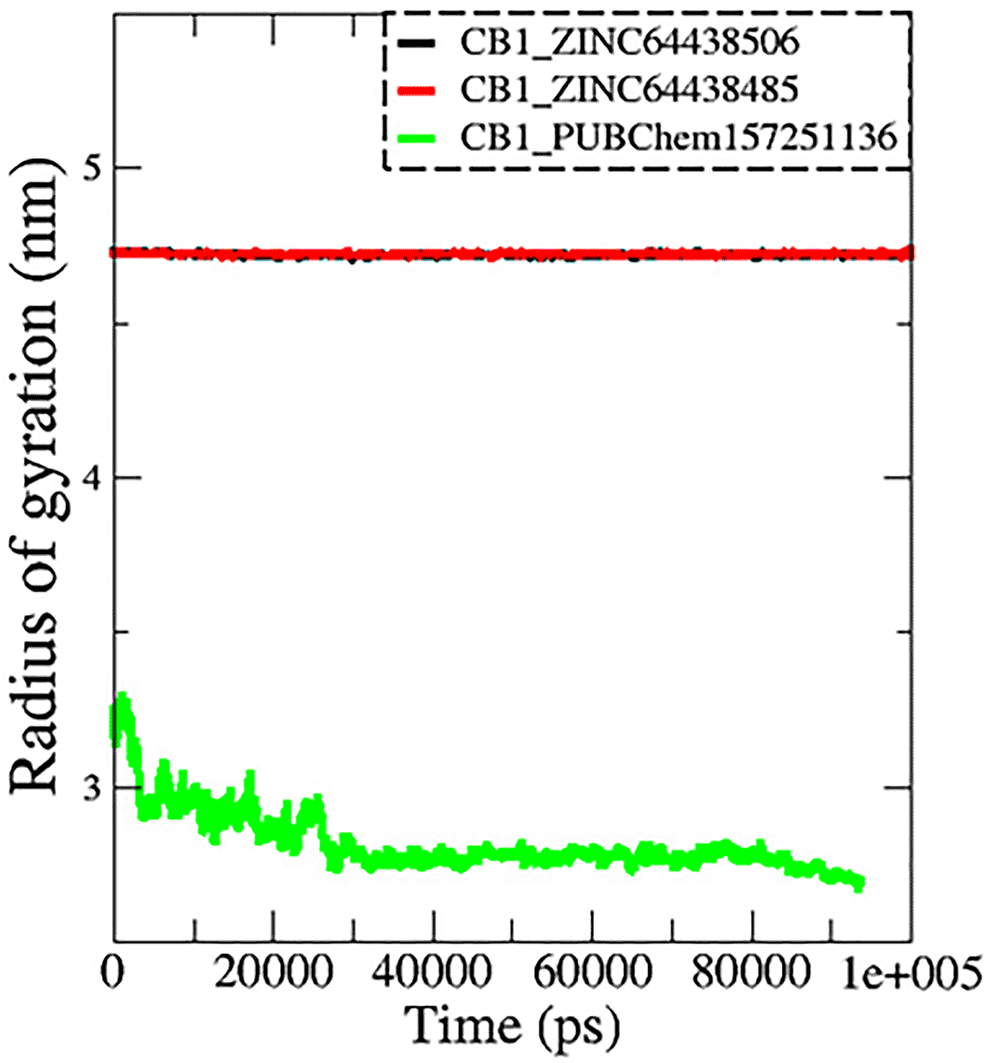
The black color represents the CB1_ZINC64438506 complex, the red color represents the CB1_ZINC64438485 complex and the green color represents the CB1_PUBChem157251136 complex.
7.4 Hydrogen bonds
One of the main factors influencing the affinity of a molecule for the protein binding pocket is its ability to form and maintain hydrogen bonds with the binding site residues. The stability of the selected agonist was assessed by analyzing the hydrogen bonds between the ligand and the protein. The results are presented in Figure 10.
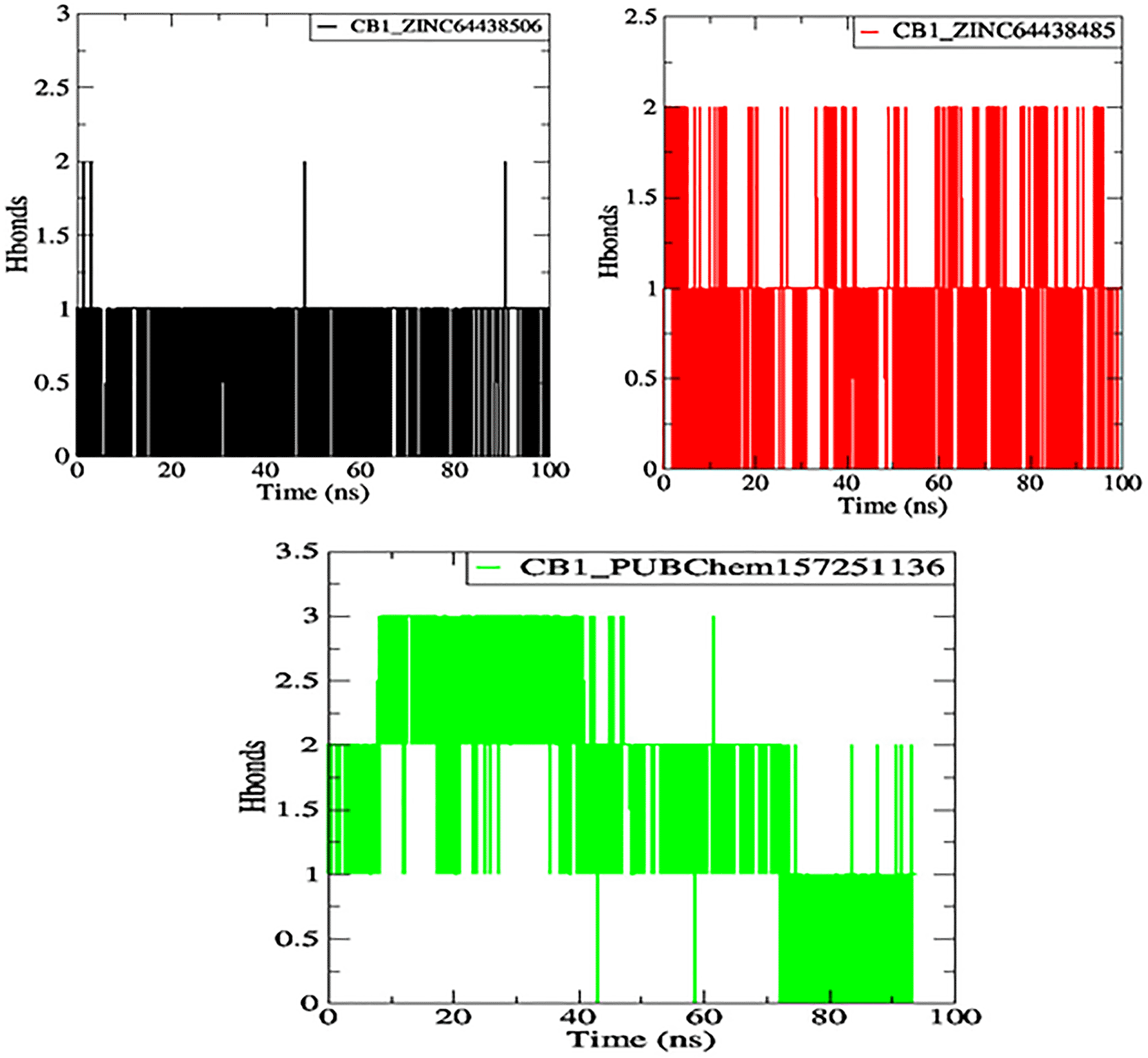
The black color represents the CB1_ZINC64438506 complex, the red color represents the CB1_ZINC64438485 complex and the green color represents the CB1_PUBChem157251136 complex.
Compounds ZINC64438506, ZINC64438485, and PUBChem157251136 form two, two and three hydrogen bonds with the CB1 binding site, respectively, indicating strong and specific interactions with the protein. These results are consistent with the docking results.
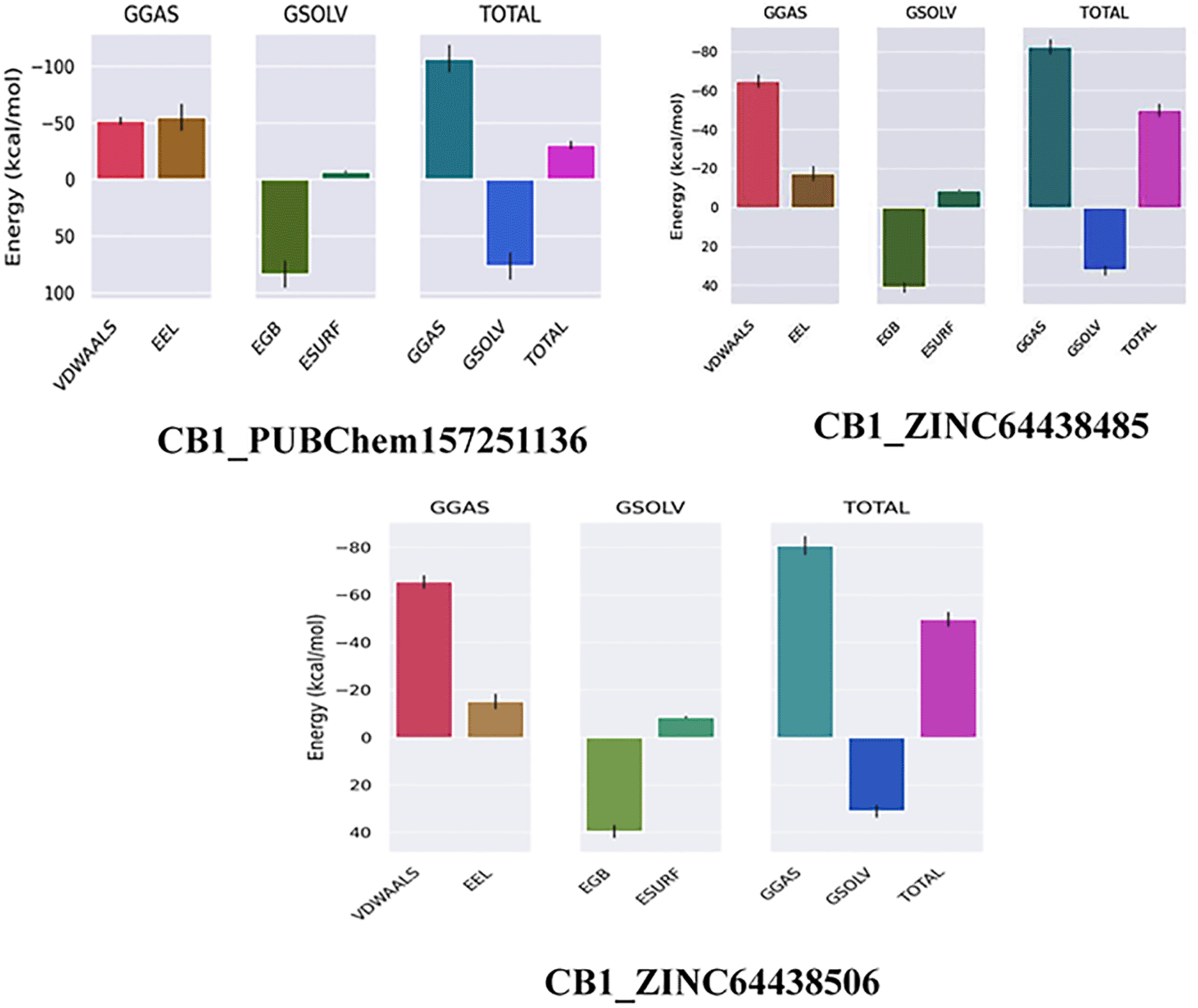
The binding free energy of all complexes was calculated to revalidate the binding affinity obtained from molecular docking analysis. The results are presented in Table 8. A ΔGbind value below -7 Kcal/mol indicates strong binding, a value between -5 and -7 kcal/mol suggests moderate binding, and a value between -2 and -5 kcal/mol corresponds to weak binding.35 The results show that all the agonists exhibit exceptionally low binding free energies, indicating a remarkable affinity for the target protein.
| Complex | ΔGGAS (Kcal/mol) | ΔGSOLV (Kcal/mol) | ΔGbind (Kcal/mol) |
|---|---|---|---|
| CB1_ZINC64438506 | -80.76 | 30.99 | -49.77 |
| CB1_PUBChem157251136 | -107.10 | 76.51 | -30.59 |
| CB1_ZINC64438485 | -82.29 | 32.31 | -49.98 |
The results also highlight the crucial role of Van der Waals and electrostatic interactions in mediating protein-ligand binding throughout the molecular dynamics simulations ( Figure 11).
Quinazoline-2,4(1H,3H)-dione derivatives represent a highly promising class of heterocyclic compounds with broad therapeutic potential, particularly as anticancer,36–38 antibacterial,39,40 antihypertensive,41 phosphodiesterase (PDE) 4 inhibition,42 5-HT3A receptor antagonist,43 anti-inflammatory,44 and anan up-and-comingtiviral agents.45 Their fused benzopyrimidinedione scaffold provides an excellent pharmacophore for targeting key biological pathways ( Figure 12). According to the article’s findings, quinazoline-2,4(1H,3H)-dione derivatives may be the first of a new class of CB1 agonists.
To synthesise the substituted quinazoline-2,4-dione and the highest-ranking agonists identified by virtual screening, namely ZINC64438506, PUBChem157251136 and ZINC64438485, several plausible synthesis strategies were developed based on established methodologies.43 These routes use various precursors such as 2-aminobenzoic acid,44 methyl 2-nitrobenzoate, 2-iodobenzamides46 or 2H-benzo [d][1,3]oxazine-2,4(1H)-dione47 ( Figure 13). The choice of starting material is guided by both its availability and synthetic feasibility. Depending on the substrate, the desired compounds can be obtained by catalytic transformations, particularly transition metal-catalysed couplings, or by intramolecular cyclisation reactions that form the characteristic quinazoline skeleton. The synthetic flexibility offered by these precursors allows the reaction conditions to be adjusted to optimise yield and purity, making them suitable candidates for further pharmacological evaluation and development.
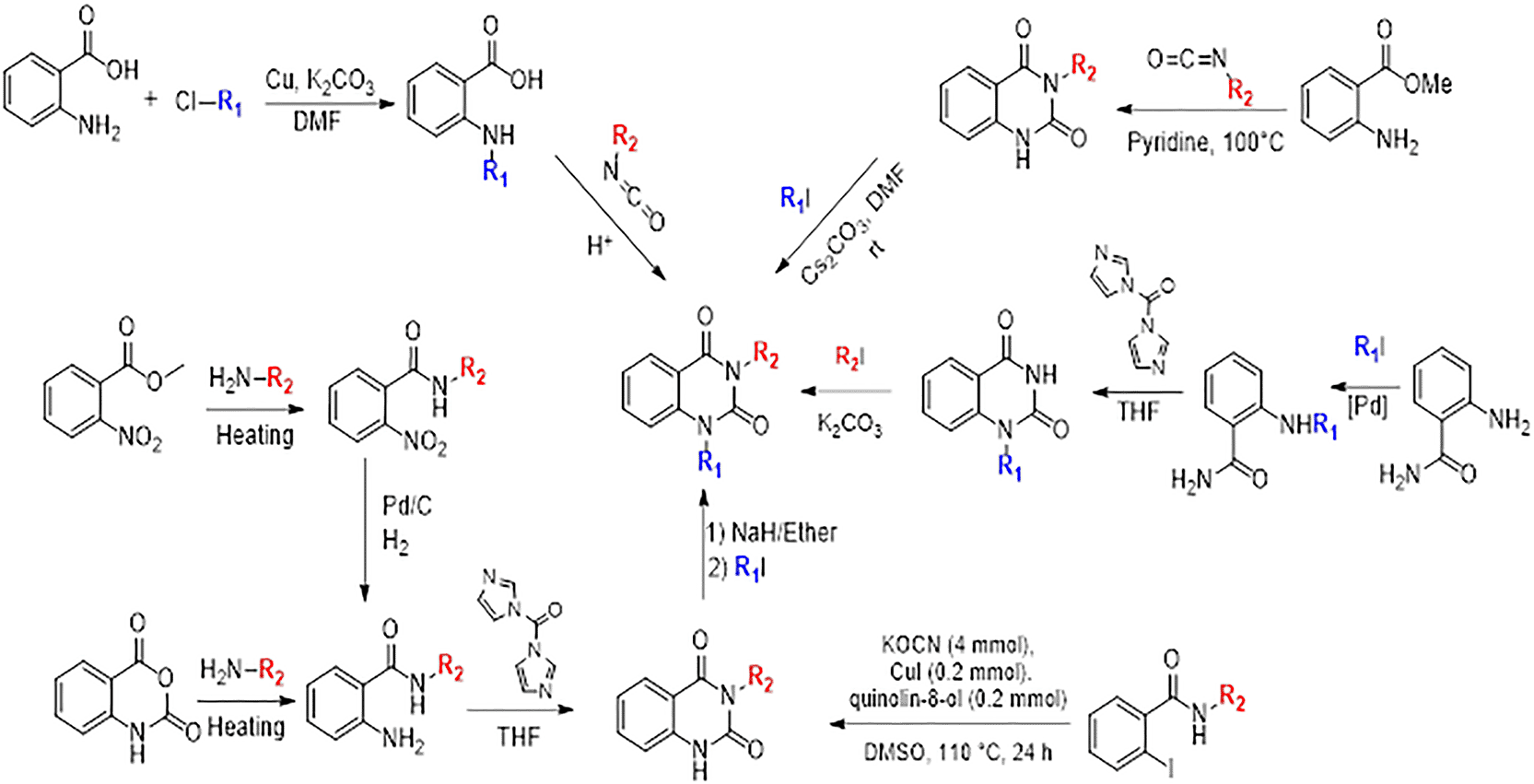
To synthesise substituted quinazoline-2,4-dione and the top-ranked agonists identified by virtual screening - namely ZINC64438506, PUBChem157251136 and ZINC64438485 - several plausible synthetic strategies have been devised, based on established methodologies.46 These routes use various precursors such as 2-aminobenzoic acid,47 methyl 2-nitrobenzoate48 which undergoes catalytic hydrogenation under mild and green conditions (1 atm of H2 in ethanol at room temperature),49,50 2-iodobenzamides51 or 2H-benzo [d][1,3]oxazine-2,4(1H)-dione52 ( Figure 13). The choice of starting material is guided by both availability and synthetic feasibility. Depending on the substrate, the desired compounds can be accessed by catalytic transformations, including transition metal-catalysed couplings, or by intramolecular cyclisation reactions that form the characteristic quinazoline scaffold. The synthetic flexibility offered by these precursors allows reaction conditions to be fine-tuned to optimize yield and purity, making them suitable candidates for further pharmacological evaluation and development.
In this study, pharmacophore-based virtual screening was conducted to identify the best CB1 agonist based on the pharmacophoric features of AM11542. The optimal pharmacophore model comprised two hydrogen bond acceptors (HBA), one aromatic ring (AR), and two hydrophobic centers (HY). This model was subsequently applied to screen a database of more than five million compounds, including Molprot with 4,742,020 molecules, ChEMBL34 with 2,264,112 molecules, ZINC with 13,127,550 molecules, ChemDiv with 1,456,120 molecules, ChemSpace with 50,181,678 molecules, Enamine with 4,117,328 molecules, MCULE with 39,843,637 molecule, MCULE-ULTIMATE with 126,471,502 molecules, NCI Open Chemical Repository with 52,237 molecules, LabNetwork with 1,794,286 molecules and PubChem with 103,302,052 molecules. Based on the generated pharmacophore model, 433 compounds were selected for further evaluation. Subsequent molecular docking refined this selection to 61 high-affinity ligands (≤-9.00 kcal/mol), which were refined to 18 promising leads through ADME-Tox analysis. Among these, three compounds were selected as potential agonists of the CB1 receptor based on their binding affinity and strong interactions with key binding pocket residues (Ser383, Ser173, His178, and Thr197). Molecular dynamics simulations using Gromacs 2024.4 confirmed the structural stability of these complexes, with low RMSD (<1 nm) and RMSF (<1 nm) values, indicating minimal conformational fluctuations. MM-GBSA calculations further validated the thermodynamic stability, with binding free energies ranging from -30.59 to -49.98 kcal/mol, reinforcing their potential as potent CB1 agonists. The three selected compounds shared a common quinazolinz-2,4(1H,3H)-dione scarfed, indicating that derivatives of this structure could pave the way for developing new CB1 receptor agonists.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)