Keywords
Amomum cardamomum, breast cancer, LC-MS, network pharmacology, molecular docking, drug discovery
This article is included in the Plant Science gateway.
Amomum cardamomum, breast cancer, LC-MS, network pharmacology, molecular docking, drug discovery
Breast cancer is one of the most common cancers in women worldwide and a leading cause of cancer-related deaths (Ye et al., 2023). According to the World Health Organization (WHO), over 2.3 million new cases of breast cancer were diagnosed globally in 2024 (World Health Organization, 2024). It remains a significant public health challenge due to its high incidence and mortality rates, particularly in low- and middle-income countries with limited access to early detection and advanced treatments (Francies et al., 2020; Manson & Achel, 2023). Despite advancements in therapeutic strategies such as chemotherapy, targeted therapy, and immunotherapy, breast cancer treatment continues to face challenges, including drug resistance, systemic toxicity, and limited efficacy in aggressive or resistant subtypes (Liu et al., 2024; Ye et al., 2023). These limitations highlight the critical need for novel therapeutic agents with improved safety and efficacy profiles.
Amomum cardamomum (Java cardamom), a member of the Zingiberaceae family, is a medicinal plant traditionally valued for its antimicrobial, anti-inflammatory, and antioxidant properties. Our previous study demonstrated that A. cardamomum exhibits significant anticancer activity against breast cancer cell lines, including its ability to inhibit cancer cell proliferation and induce apoptosis (Khairani et al., 2024). These findings provided strong preliminary evidence of A. cardamomum’s potential as a therapeutic agent for breast cancer. Building on this foundation, the present study aims to further explore the therapeutic potential of A. cardamomum by elucidating the bioactive compounds responsible for its anticancer effects and their underlying mechanisms of action. Using Liquid Chromatography-Mass Spectrometry (LC-MS), we identified and characterized the phytochemicals present in A. cardamom. A network pharmacology approach was employed to identify key protein targets and pathways associated with these compounds to complement this. Molecular docking studies and molecular dynamics simulations assessed these compounds’ binding interactions and stability with their targets. Additionally, ADMET profiling and toxicity assessments were conducted to evaluate the drug-likeness and safety of the identified compounds.
This study represents a critical step in advancing the understanding of A. cardamomum’s bioactive potential and its application in addressing the challenges of breast cancer therapy. By integrating in vitro findings, compound characterization, and computational approaches, we aim to provide insights that could contribute to developing novel, plant-based therapeutics for breast cancer treatment.
This study employed a multi-faceted methodological framework to comprehensively evaluate the therapeutic potential of A. cardamomum for cancer treatment. The methods included LC-MS analysis for compound identification, network pharmacology to identify key protein targets, molecular docking and dynamics to analyze ligand-target interactions, and ADMET profiling and toxicity assessments to evaluate drug-likeness and safety. Each method was selected to address specific aspects of the study objectives and to ensure a thorough investigation of A. cardamomum’s bioactive compounds.
2.1.1 Liquid Chromatography-Mass Spectrometry (LC-MS) analysis
The ethanol extract of Java cardamom seeds, which already extracted in our previous study, was analyzed using a UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm) on an LC-MS system operating in positive ion mode. A sample volume of 5 μL was injected into the system for each run. The extract was prepared by dissolving 100 μL of the liquid extract in 900 μL of water, vortexed, and filtered through a 0.22 μm Millex filter before injection. The mobile phase consisted of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). The separation was performed using gradient elution with the following profile: 95% A and 5% B at 0.4 minutes, gradually transitioning to 100% B over 15 minutes. The composition was held at 100% B for 0.1 minutes before returning to the initial condition of 95% A and 5% B at 15.1 minutes, which was maintained until 20 minutes for re-equilibration. The flow rate was set to 0.4 mL/min. Mass spectrometry was detected using electrospray ionization (ESI) in positive ion mode, with a mass range of 50–1200 m/z. The capillary voltage was set to 0.5 kV, the cone voltage to 21 V, and the source temperature to 500°C (Abdallah et al., 2023; Kaur et al., 2022).
2.2.1 Protein-protein interaction and protein network construction
Protein-protein interaction (PPI) networks were constructed by integrating overlapping protein targets associated with breast cancer, utilizing multiple databases for comprehensive analysis. The process involves querying GeneCards and the Chemical Toxicogenomic Database using the search term “breast cancer” while incorporating predicted protein targets of cardamom compounds obtained from SwissTarget Prediction. The intersection of these protein sets is then input into the STRING database, specifically focusing on Homo sapiens with a medium confidence threshold (score > 0.4). The resulting protein network is subsequently imported into Cytoscape, a network visualization software, for in-depth analysis and more representative visualization of the protein interactions (Hozhabri et al., 2022). This approach enables researchers to elucidate complex relationships between proteins involved in breast cancer and potential therapeutic targets, providing valuable insights into the molecular mechanisms underlying the disease. By leveraging multiple databases and sophisticated visualization tools, scientists can identify key nodes and clusters within the PPI network, potentially uncovering novel therapeutic targets or biomarkers for breast cancer.
2.2.2 Protein and ligand preparation
Four selected proteins from the PPI network result were obtained from the PDB website (https://www.rcsb.org/; accessed November 14th, 2024) ( Table 1). The Biovia Discovery Studio v.2021 application was used to separate the control ligand from the three-dimensional crystal structure of the target protein. The two-dimensional structure of A. cardamomum’s bioactive compounds was retrieved from the PubChem website in SDF format (https://pubchem.ncbi.nlm.nih.gov/; accessed November 14th, 2024). The MOE v2022.02 application was then used to conserve, neutralize, and refine all ligands and proteins to an RMS gradient of 0.001 kcal/mol/Å2 (Yenny et al., 2024).
2.2.3 Pharmacokinetic and pharmacodynamic analysis of A.cardamomum compounds
Assessing a compound’s absorption, distribution, metabolism, excretion, and toxicity (ADMET) characteristics requires multiple computational methods. First, the canonical SMILES notation of the compound was obtained from the PubChem Database. The canonical SMILES notation of the compounds was then inserted into the SwissADME server (http://www.swissadme.ch/index.php; accessed November 15th, 2024) to determine the ADME (absorption, distribution, metabolism, and excretion) properties. Furthermore, the toxicity predictions of all the compounds were conducted using the ProTox-II server (http://tox-new.charite.de/protox_II; accessed November 15th, 2024) in compliance with the toxicity criteria set by the Organisation for Economic Cooperation and Development (OECD). The drug-likeness of the compound was assessed using Lipinski’s rule of five criteria. The compounds were evaluated for their adherence to Lipinski’s principles by submitting their canonical SMILE molecular structures to the Molinspiration Cheminformatics service (https://www.molinspiration.com/cgi/properties; accessed November 15th, 2024).
2.2.4 Molecular docking studies
The MOE v2022.02 application was used to perform the molecular docking. MOE’s Site Finder tool automatically finds the amino acid sequences of the target protein’s active site ( Table 1). The results of the molecular docking include 2D/3D visualization, Root Mean Square Deviation (RMSD) (Å), and binding affinity (kcal/mol). Compounds are regarded as potent target protein inhibitors if their RMSD value is less than 2Å and their binding affinity is much lower (more negative value) than the control ligand (Yenny et al., 2024). Additionally, the type and similarity of interaction between the chosen compound and the control ligand in the target protein’s active site were used to visualize and ensure that the compound could act as a specific inhibitor of the target protein (Rita et al., 2025).
2.2.5 Molecular dynamic simulation
The ligand with the lowest binding affinity was further analyzed through molecular dynamics simulation using Yet Another Scientific Artificial Reality Application (YASARA) software (Krieger & Vriend, 2015). The simulation parameters were set at 25°C temperature, pH 7.4, and 0.9% salt concentration for a duration of 50 nanoseconds. This designated timeframe significantly exceeds the minimum duration required to assess protein-ligand complex stability, which is typically 20 nanoseconds (Wargasetia et al., 2021). The simulation was executed using the md_run macro program, and the results were analyzed using the md_analyze function.
3.1.1 Liquid Chromatography-Mass Spectrometry (LC-MS) analysis
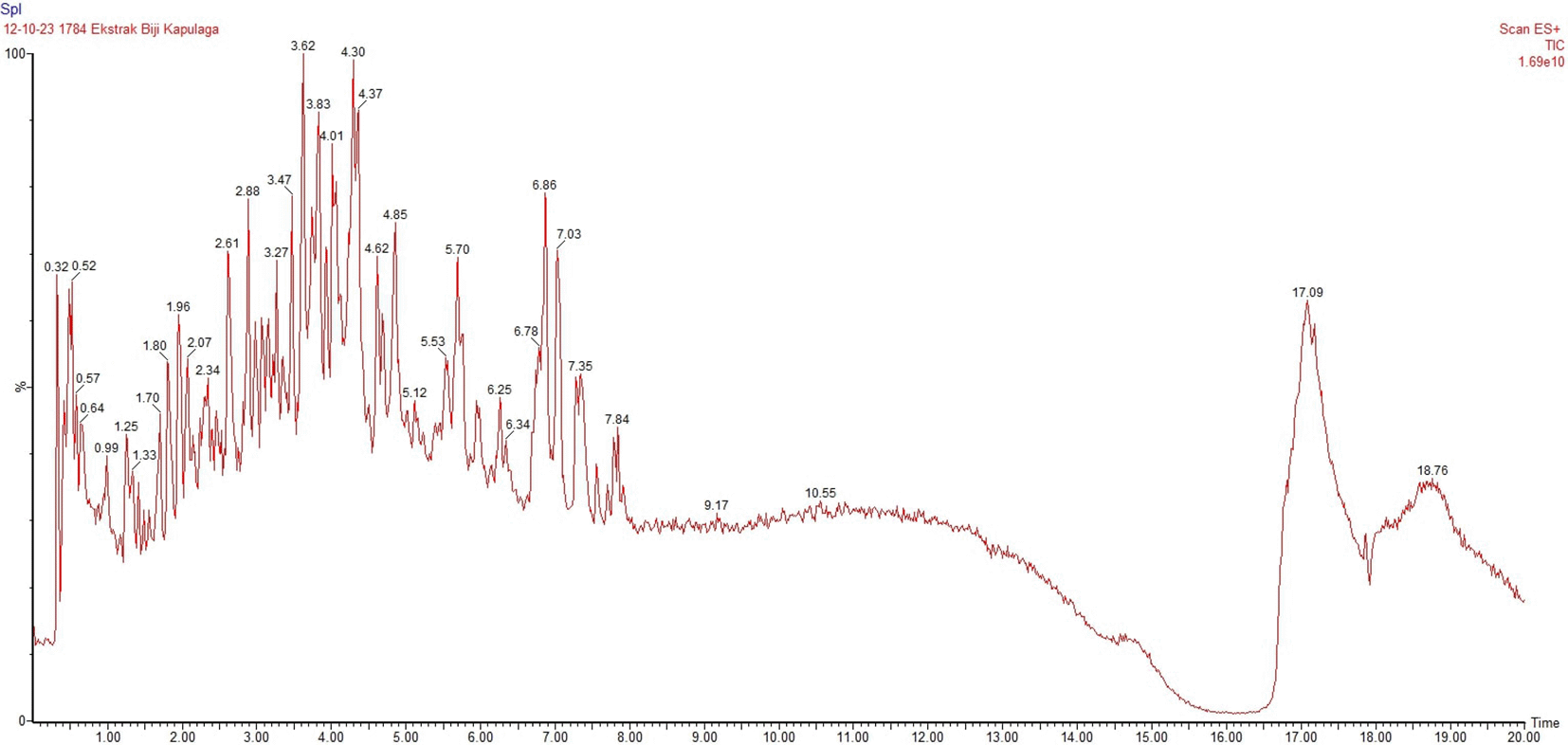
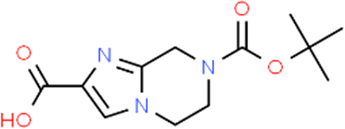
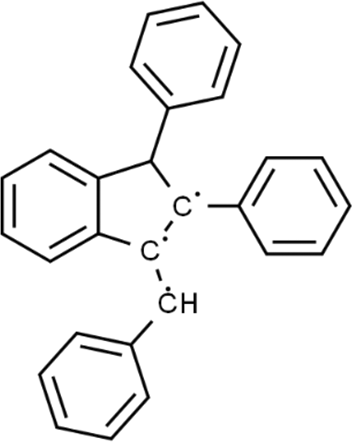
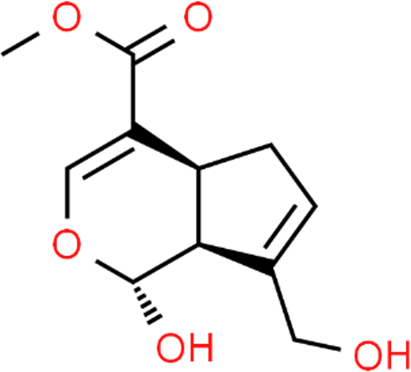
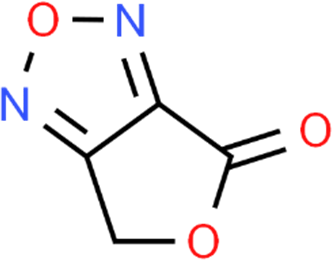
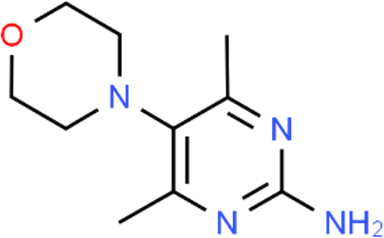
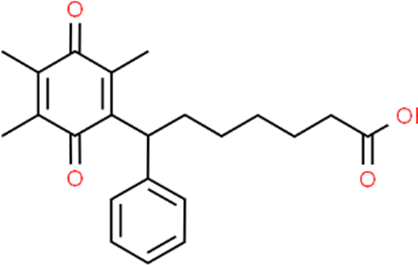
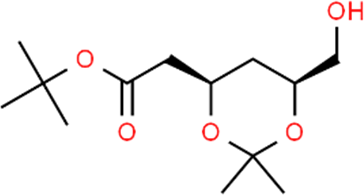
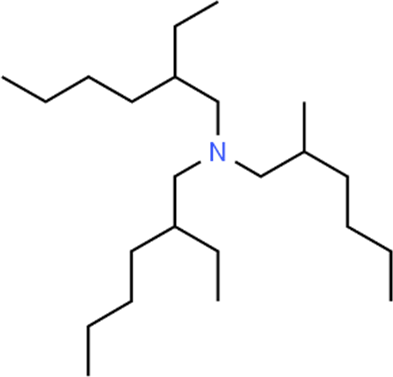
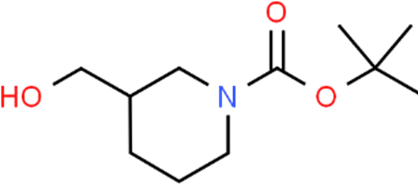
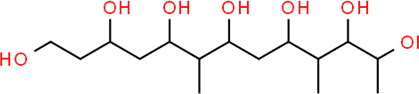
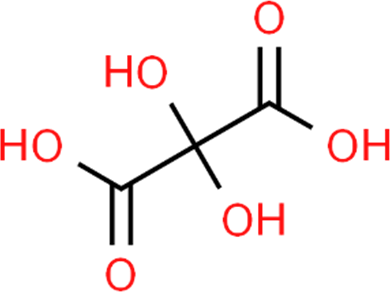
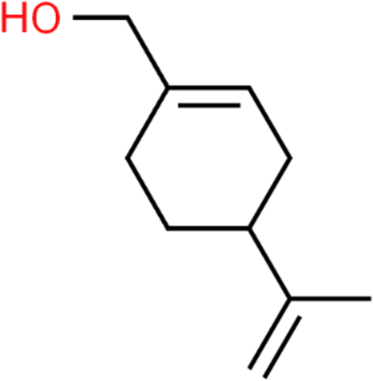
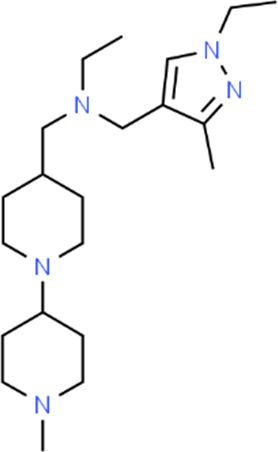
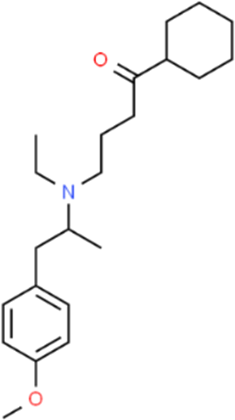
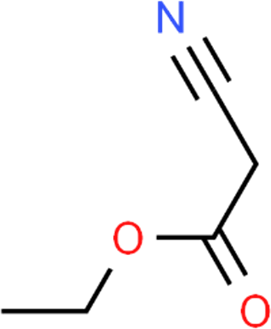
The LC-MS analysis of Amomum cardamomum seed extract identified 22 unique compounds ( Figure 1, Table 2). These compounds exhibit diverse molecular weights, retention times, and polarities, reflecting the chemical complexity of the extract. The retention times ranged from 0.52 to 17.09 minutes, and molecular weights varied between 113.115 Da (ethyl cyanoacetate) and 369.161 Da (1,2-diphenyl-3-(phenylmethyl)-1H-indene), highlighting the presence of both small and larger molecules.
3.2.1 Protein-protein interaction network analysis
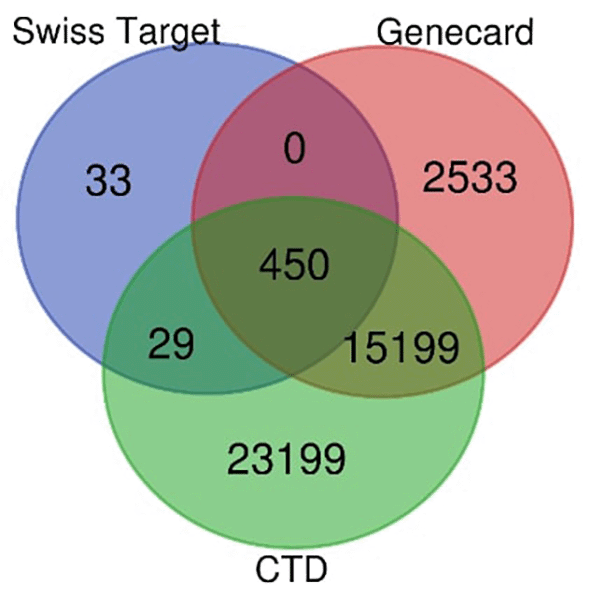
The analysis of 22 compounds from cardamom extract revealed 512 potential targets based on Swiss target prediction. These results were compared with breast cancer-related genes from GeneCards (18,182 genes) and CTD (38,877 genes). A Venn diagram showed ( Figure 2.) 450 genes at the intersection of breast cancer and bioactive compounds from A. cardamomum, representing the overlap between cardamom-specific targets and breast cancer-related genes.
To investigate the hub genes within key modules, a protein-protein interaction (PPI) network analysis was conducted using STRING with a confidence score ≥0.4 for the biological species “Homo sapiens” ( Figure 3). This analysis revealed the synergistic and multi-targeted effects of A. compacta, contributing to its potential anticancer properties. As shown in Figure 2, topology analysis using Cytoscape was performed to calculate the centrality of each protein. The study identified CAS3, EGFR, SRC, and TNF-18 as key targets with the highest degree of connectivity, indicating their essential roles in the mechanisms of breast cancer ( Figure 3). These findings are supported by previous research highlighting the importance of these proteins in breast cancer progression and treatment resistance (Hozhabri et al., 2022).
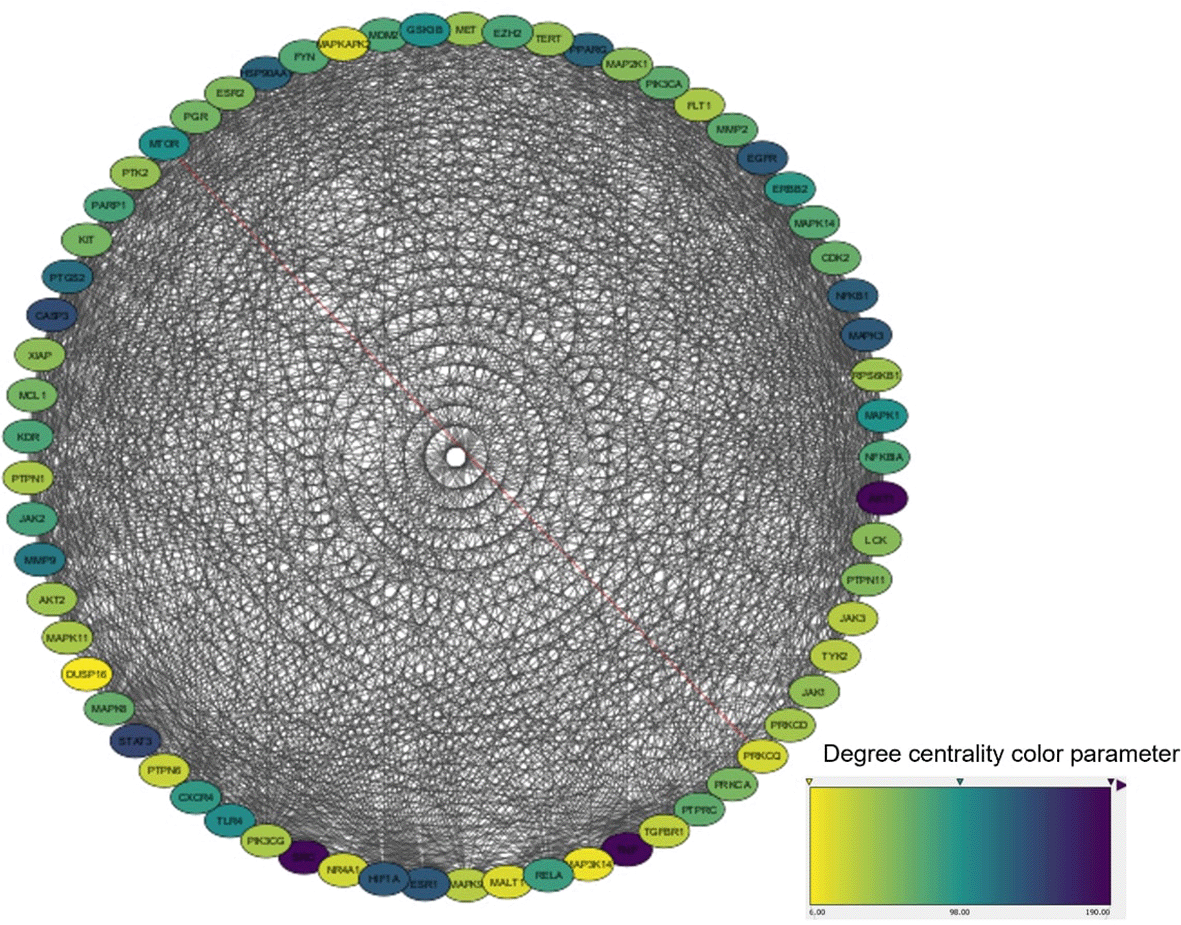
The high centrality of these proteins in the PPI network suggests their critical roles in the complex interplay of molecular pathways involved in breast cancer. Their identification as key targets provide valuable insights into potential therapeutic strategies. Consequently, these proteins were selected as ideal candidates for further analysis through molecular docking and molecular dynamics simulations, which can offer a deeper understanding of their interactions with potential anticancer compounds and guide the development of more effective treatments for breast cancer (Kanhaiya et al., 2017).
3.2.2 Pharmacokinetic and pharmacodynamic analysis of A. cardamomum compound
The pharmacokinetic and pharmacodynamic analysis of the 22 compounds identified in A. cardamomum seed extract revealed several key findings regarding their drug-likeness and potential therapeutic application ( Table 3).
3.2.2.1 Absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis
The pharmacokinetic evaluation ( Table 3) revealed that most compounds from A. cardamomum seeds were predicted to have good gastrointestinal absorption, suggesting favorable uptake following oral administration. Only a few compounds, including C5, C7, C15, and C18, showed low absorption potential, indicating possible limitations in bioavailability. Consistent with these findings, nearly all compounds exhibited adequate oral bioavailability, reinforcing their potential for systemic circulation. Interestingly, several molecules, such as C1, C3, C11-C14, C16, C19-C21, were also predicted to penetrate the blood–brain barrier, raising the possibility of central nervous system activity.
In terms of metabolism, the majority of compounds were not identified as substrates or inhibitors of cytochrome P450 2D6 (CYP2D6), which reduces the likelihood of extensive enzyme-mediated clearance or drug–drug interactions. Notably, C15 was predicted to act as a substrate, while C21 displayed inhibitory potential, highlighting that these particular molecules may influence or be influenced by drugs processed through CYP2D6.
Excretion analysis indicated that almost all compounds were unlikely to interfere with renal elimination pathways. Only C15 demonstrated potential inhibitory activity toward the organic cation transporter OCT2, suggesting that most A. cardamomum constituents are unlikely to alter the renal clearance of co-administered drugs.
Regarding solubility, the majority of compounds showed adequate water solubility, ranging from moderate to very soluble, which is advantageous for formulation and bioavailability. However, two compounds, C6 and C15, were categorized as poorly soluble, which could limit their practical application unless addressed by formulation strategies. Together, these findings suggest that the chemical profile of A. cardamomum seeds is largely consistent with drug-likeness criteria, although specific molecules may require optimization to overcome solubility or metabolic limitations.
3.2.2.2 Lipinski’s rules of five
Lipinski’s rule of five indicates that 20 of the 22 evaluated compounds meet the criteria for drug-likeness compounds (Table. 4). However, two compounds, C15 and C18, do not fully adhere to these criteria. C15 exhibits noncompliance due to the consensus log P value >5 (6.964573) and molar refractivity >130 (130.880981), which indicates potential challenges related to excessive lipophilicity and molecular size. LogP value indicates the level of lipophilicity, where high lipophilicity can result in quickly metabolized compounds, low solubility, and poorly absorbed (Rutkowska et al., 2013). Molar refractivity values within the range of 40-130 suggest that a substance is likely to have good intestinal absorption and oral bioavailability (Ya’u Ibrahim et al., 2020). Conversely, C18 fails to meet the criteria because of its H-bond receptor count >5 (6) and molar refractivity <40 (17.236599), indicating potential issues with polarity and molecular size. The H-bond acceptor and H-bond donor present in the structure of a therapeutic agent play a vital role in membrane transport, drug-protein interactions, distribution, and aqueous solubility (Kenny, 2022). This evaluation provides valuable insights into the drug-like potential of the compounds from A. cardamomum.
3.2.2.3 Toxicity prediction
Based on the results of the toxicity analysis ( Table 5), three compounds, namely C6, C7, and C14, were found to be in the toxic level (class I-III). Overall, one compound (C14) is classified as class two toxicity (fatal by ingestion (5 < LD50 ≤ 50)), two compounds (C6 and C7) are classified as class III toxicity (toxic by ingestion (50 < LD50 ≤ 300)), twelve compounds (C1, C2, C4, C5, C8, C9, C11, C13, C15, C16, C20, and C21) are classified as class IV toxicity (harmful if swallowed (300 < LD50 ≤ 2000)), six compounds (C3, C10, C17, C18, C19, and C22) are classified as class V toxicity (may be harmful if swallowed (2000 < LD50 ≤ 5000)), and one compound (C12) are classified as class VI toxicity (non-toxic (LD50 > 5000)).
Based on the analysis of organ toxicity, only 1 out of 22 compounds (C2) was reported to be active in the hepatotoxicity category. In the next category, 7 compounds (C4, C5, C6, C8, C10, C12, and C22) were reported as active carcinogenicity. Two out of 22 compounds (C4 and C8) were reportedly active in the mutagenicity. All evaluated compounds did not exhibit immunotoxicity or cytotoxicity. This is a positive sign that these compounds may not always risk genetic or cell damage, even if they are considered detrimental or possibly hazardous based on LD50 values [3]. Therefore, even if the chemicals from A. cardamomum have a variety of toxicity profiles, any prospective medicinal or pharmacological application must consider the unique risk factors associated with each component. Moreover, in vivo investigations are necessary to validate these in silico results and offer a more thorough comprehension of the safety and effectiveness of these substances.
3.2.3 Molecular docking
The molecular docking analysis was conducted using the MOE application. To carry out a targeted molecular docking analysis, the grid box is accurately placed at the active site of each target protein. The grid box settings in the MOE application’s Site Finder feature were automatically modified according to the amino acid residues corresponding to the natural ligand of each protein. Molecular docking results include binding affinity (measured in kcal/mol), RMSD (Å), and visualization in two and three dimensions. A peptide with an RMSD below 2.5 Å and a significantly higher binding affinity (more negative) than the control ligand may be regarded as a promising inhibitor of the target protein.
3.2.3.1 Molecular docking of SRC protein
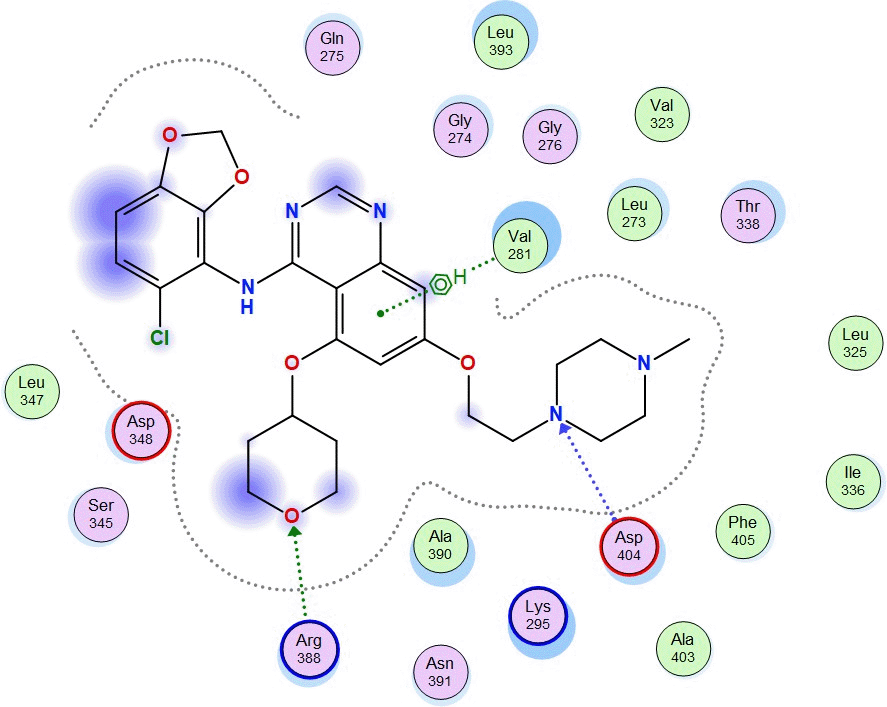
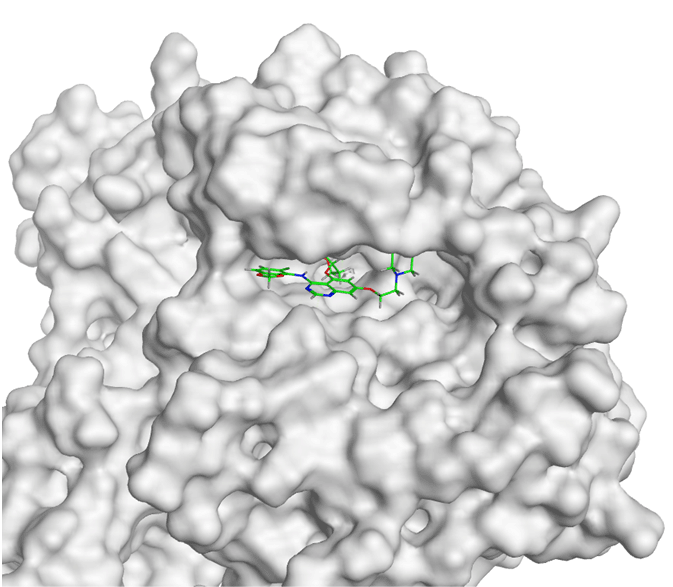
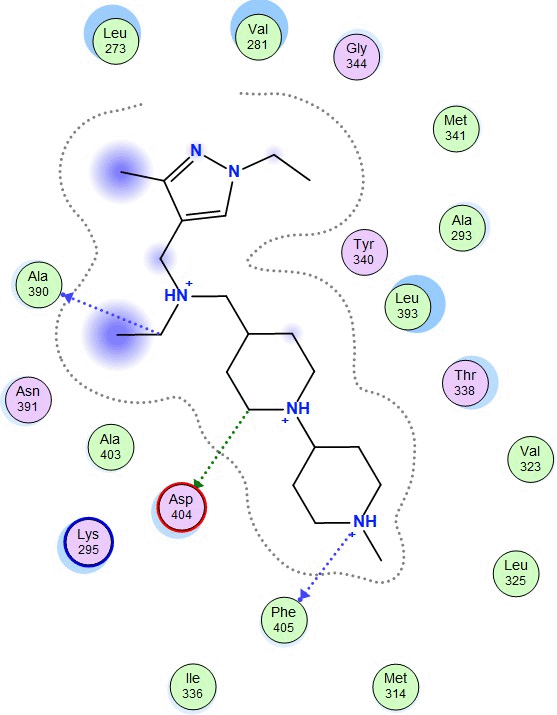
The molecular docking between SRC and Amomum cardamomum revealed that the compounds of Amomum cardamomum have RMSD values below 2Å. Based on the binding affinity, C20 has the lowest binding affinity (-8.58 kcal/mol), stronger than the control ligand, with a binding energy of -7.89 kcal/mol ( Table 6). C20 formed three interactions: one acidic hydrophilic (Asp404) and two greasy hydrophobic interactions (Ala390 and Phe405). Control ligand formed three interactions: one acidic hydrophilic (Asp404), one basic hydrophilic (Arg388), and one greasy hydrophobic interaction (Val281). These results showed that C20 interacts with Asp404, the same amino acid residue with which the control ligand interacts ( Table 7). It can be concluded that C20 has the potential to be a new specific competitive inhibitor of SRC active site.
3.2.3.2 Molecular docking of TNF-α protein
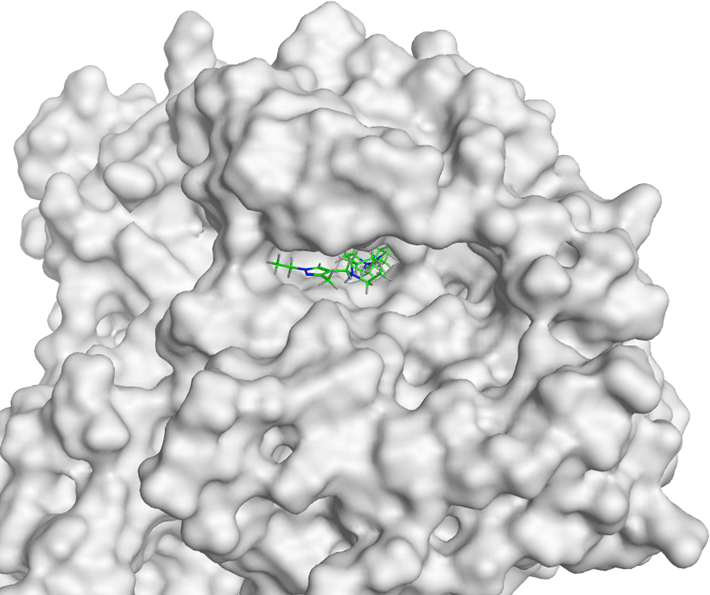
From the molecular docking result, C17 attaches to the TNF-α active site with minimal deviation, marked by RMSD < 2Å (1.41 Å). C17 (-6.91 kcal/mol) has a binding affinity close to the control ligand’s (-7.00 kcal/mol) ( Table 6). C17 formed three polar hydrophilic interactions: TyrC119, TyrA119 and TyrA151. Meanwhile, the control ligand formed two polar hydrophilic interactions: TyrA119 and TyrA151. These showed that C17 and the control ligand interact with two similar amino acid residues of TNF-α (TyrA119 and TyrA151) ( Table 8). So, C17 has the potential as a new specific competitive inhibitor of TNF-α.
3.2.3.3 Molecular docking of Casp3 protein
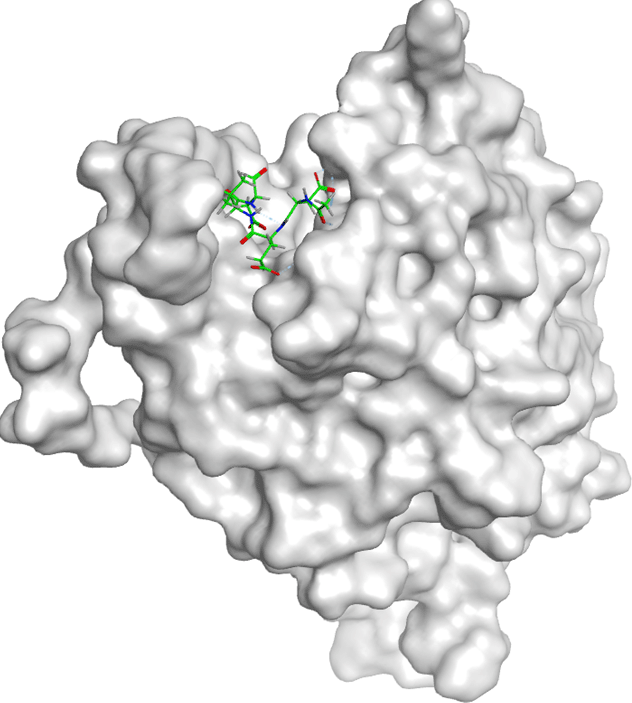
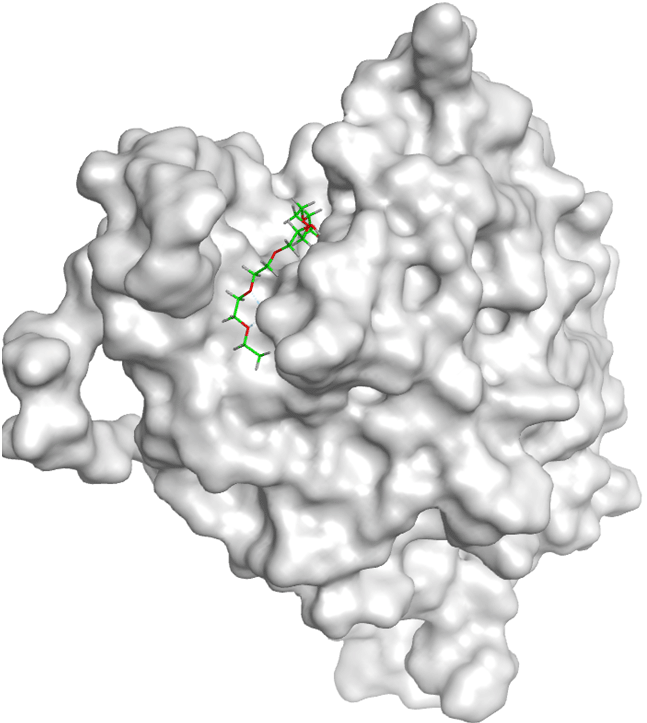
All compounds of Amomum cardamomum have an RMSD value < 2Å. Based on the binding affinity, C17 has the highest binding affinity than the other compounds of A. (-6.70 kcal/mol) cardamomum and drug control ( Table 6). The 2D interaction visualization revealed that C17 formed three interactions with the active site of casp3: one basic hydrophilic (Arg207), one polar hydrophilic (Asn208), and one greasy hydrophobic (Trp214) interaction. On the other hand, casp3 inhibitors formed one basic hydrophilic (Arg207) and two polar hydrophilic interactions (Thr62, Ser209) with the active site of casp3. This result showed that C17 and casp3 inhibitors formed one similar interaction with the active site of casp3 (Arg207) ( Table 9). In conclusion, C17 has the potential as a new specific competitive inhibitor of the casp3 active site.
| Ligand | 2D Interaction | 3D Interaction |
|---|---|---|
| Control Drug | 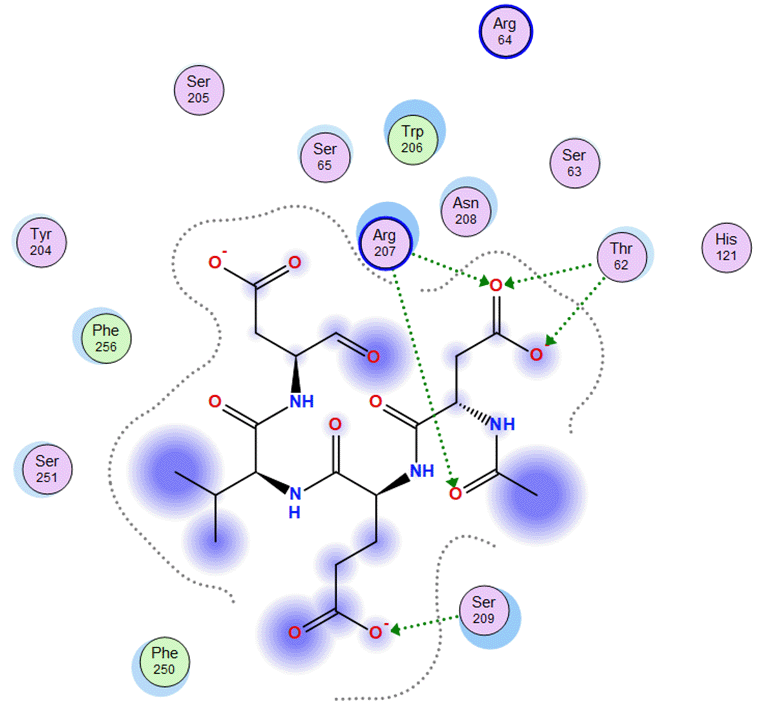
| 
|
| C17 | 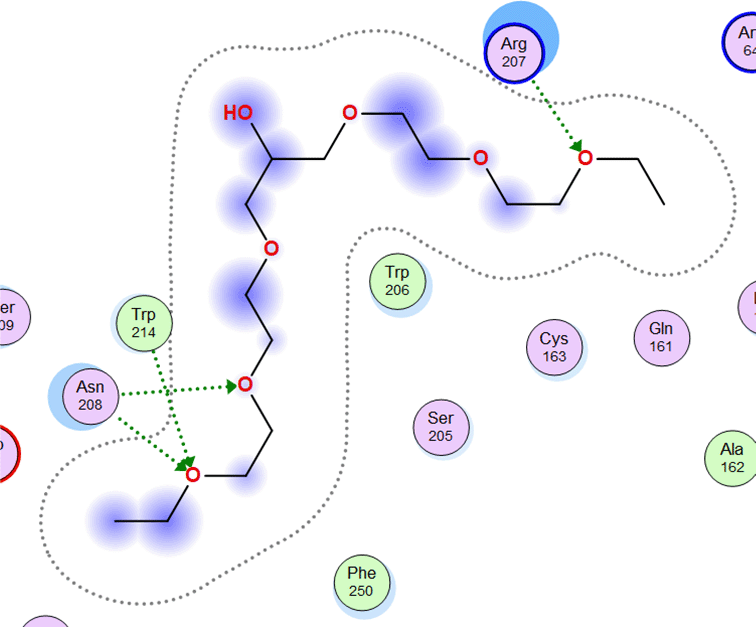
| 
|
3.2.3.4 Molecular docking of EGFR protein
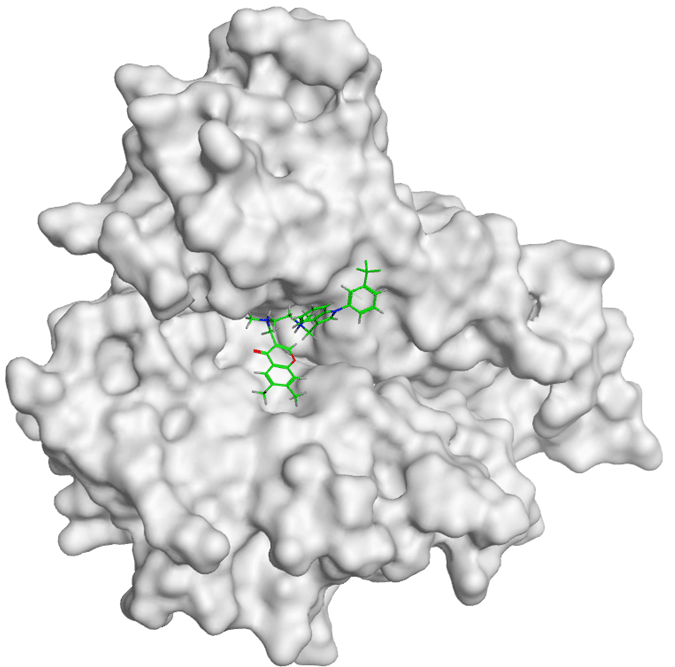
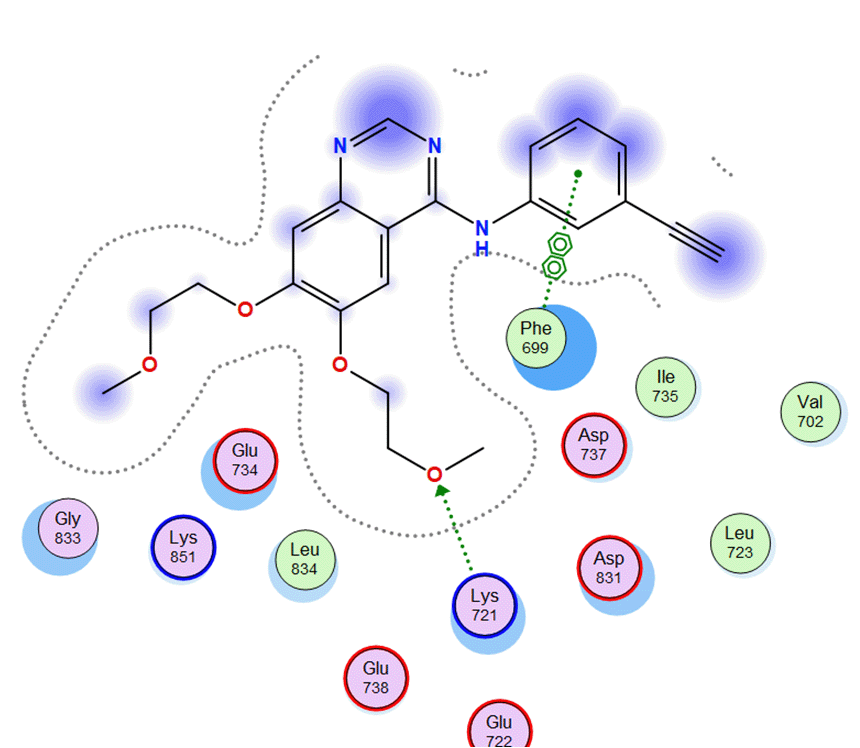
All compounds of Amomum cardamomum have RMSD values below 2 Å, except C15. Regarding binding affinity, C20 (-6.43 kcal/mol) has a binding affinity close to the control ligand (-6.47 kcal/mol) as the control ligand ( Table 6). Although the binding affinity of C20 is not stronger than the control ligand, the difference in binding affinity is insignificant. The result of 2D interaction visualization showed that C21 formed two acidic hydrophilic interactions (Asp831 and Glu 738) with the active site of EGFR. Meanwhile, the control ligand formed one basic hydrophilic interaction (Lys721) and one greasy hydrophobic interaction (Phe699). Although C20 does not have identical amino acid residues as the control ligand, the 3D interaction visualization results show that C20 can occupy the binding site of the control ligand on the EGFR active site ( Table 10).
3.2.4 Molecular dynamic simulation
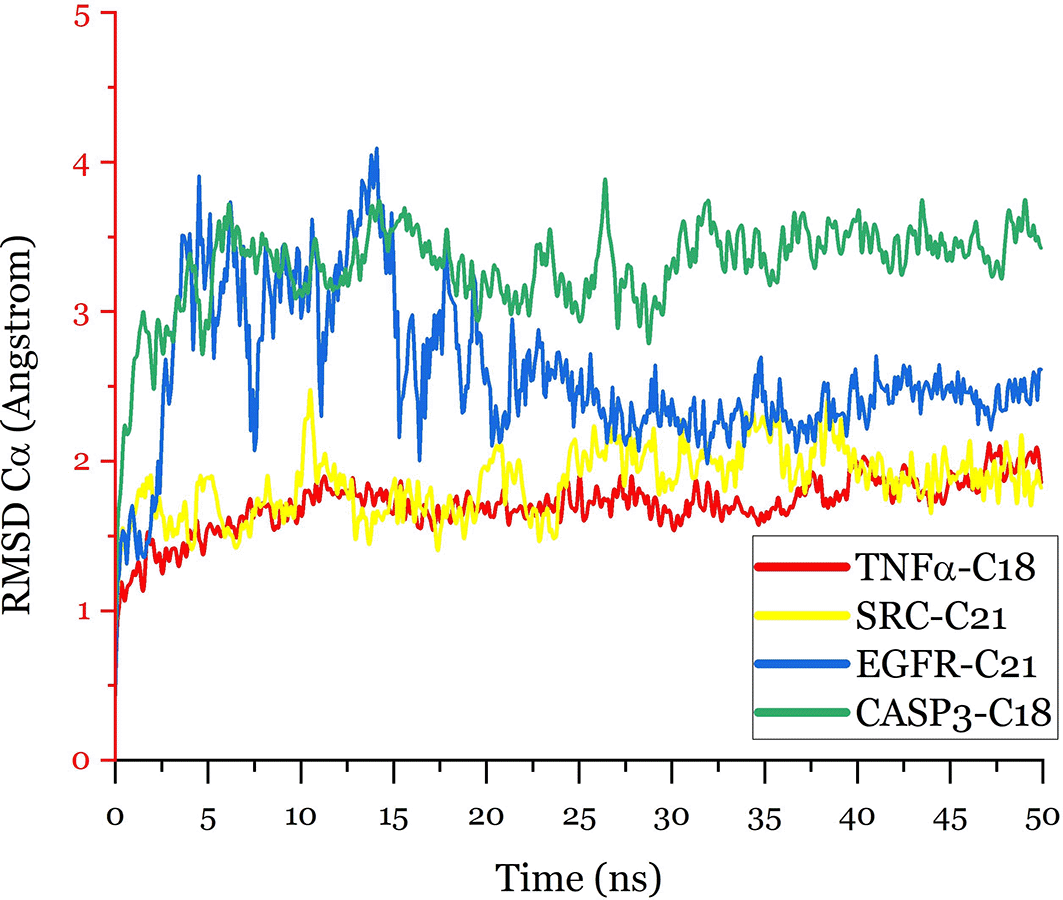
The graph illustrates that TNFα-C18 exhibits stable behavior, with RMSD values consistently around 1.5-2.0 Angstroms during the 50 nanoseconds simulation. SRC-C21 shows similar stability with RMSD fluctuating around 1.5-2.0 Angstroms, though with slightly more variation than TNFα-C18. EGFR-C21 demonstrates higher RMSD values (2.5-4.0 Angstroms) with significant fluctuations, particularly in the first 25 nanoseconds, before stabilizing around 2.5 Angstroms. CASP3-C18 shows the highest average RMSD values, maintaining levels between 3.0-3.5 Angstroms throughout the simulation with notable fluctuations. All molecular complexes reach relative stability after approximately 25 nanoseconds, though with different characteristic RMSD ranges ( Figure 4).
This study demonstrated the therapeutic potential of A. cardamomum through a combination of LC-MS analysis, pharmacokinetic and pharmacodynamic prediction, network pharmacology, molecular docking, and molecular dynamic simulation. LC-MS analysis identified 22 unique compounds in the seed extract, showcasing a chemically diverse profile with various molecular weights and retention times. The protein-protein interaction (PPI) network studies revealed key protein targets for A. cardamomum compounds, including SRC, EGFR, TNF-α, and Casp3, all of which play critical roles in breast cancer progression.
SRC, a non-receptor tyrosine kinase and the first proto-oncogene identified in mammalian cells plays a crucial role in breast cancer (BC) by promoting tumor initiation, growth, metastasis, and drug resistance (Luo et al., 2022). SRC is overexpressed or activated in various stages of BC, where it enhances signaling pathways that promote cell proliferation, survival, and motility, contributing to tumorigenesis. SRC activation leads to the phosphorylation of epidermal growth factor receptor (EGFR) (Finn, 2008; Pelaz & Tabernero, 2022; Raji et al., 2024; Wheeler et al., 2009). Additionally, SRC interacts with HER2, amplifying signaling through pathways like FAK and STAT3, which increase the metastatic potential and contribute to resistance against HER2-targeted therapies (Raji et al., 2024). Inhibition of SRC has been shown to disrupt these pathways and reduce tumorigenesis, making SRC a critical target for therapeutic intervention in BC. One compound that can inhibit SRC activity is Quinazoline. Quinazoline exerts its effects by binding to the ATP-binding pocket of SRC, leading to the inhibition of its kinase activity. This interaction disrupts signaling pathways essential for tumor cell survival and growth, such as the PI3K-AKT and MAPK pathways (Emami et al., 2022).
Tumor necrosis factor (TNF-α) is a pro-inflammatory cytokine that plays a significant role in developing and progressing breast cancer. TNF-α is involved in carcinogenesis by activating several signaling pathways, including the TNF receptor 1 (TNFR1) pathway, which leads to the upregulation of oncoproteins such as HBXIP. This activation promotes cell proliferation, survival, and migration, contributing to tumorigenesis. Elevated TNF-α levels are often observed in breast cancer tissues, and its signaling is linked to increased metastasis and drug resistance, making TNF-α a critical factor in BC progression. Inhibition of TNF-α has been shown to reduce cancer cell proliferation, suppress tumor growth, and decrease metastatic potential. Consequently, TNF-α inhibitors are being explored as potential therapeutic agents for BC treatment. Targeting TNF-α makes it possible to disrupt its downstream signaling pathways, including the NF-κB and MAPK pathways, crucial for maintaining tumor cell survival and inducing inflammatory responses that support tumor growth (Cai et al., 2017; Mercogliano et al., 2020). Given the importance of TNF-α in breast cancer progression, targeting its activity offers a promising strategy to mitigate carcinogenesis and enhance the efficacy of existing therapies.
Caspase-3 (Casp3) plays a complex role in breast cancer, acting as a key apoptosis regulator, but its dysregulation can contribute to tumor progression and chemotherapy resistance (Pu et al., 2017). High Casp3 expression is associated with poor prognosis, particularly in HER2-positive tumors and specific patient populations, while Casp3 deficiency reduces tumorigenesis in preclinical models (Pu et al., 2017; Zhu et al., 2024). Casp3 activation is crucial for apoptosis, but its activity can be inhibited by factors such as the X-linked inhibitor of apoptosis protein (XIAP), which prevents its activation and subsequent cell death (Yosefzon et al., 2018). These findings highlight Casp3’s dual role in promoting tumorigenesis and resistance to treatment, suggesting it could be a target for therapeutic strategies in breast cancer.
Epidermal growth factor receptor (EGFR) is a key player in breast cancer progression, as it regulates critical processes like cell proliferation, survival, migration, and differentiation. EGFR overexpression in breast cancer leads to the dysregulation of signaling pathways such as PI3K-AKT and MAPK, promoting tumorigenesis, metastasis, and resistance to apoptosis (Sigismund et al., 2017). This is particularly evident in triple-negative breast cancer (TNBC), where EGFR overexpression is linked to poor prognosis. EGFR also interacts with other receptors, like HER2, amplifying tumor growth signals (Masuda et al., 2012). Due to its central role, EGFR is potentially to be a target for therapeutic strategies
The integration of molecular docking and dynamic simulation analyses provided robust evidence for the therapeutic potential of A. cardamomum compounds, mainly C17 and C20, in targeting critical proteins involved in breast cancer. These compounds demonstrated strong binding affinities and maintained stable interactions with their targets during simulations, underscoring their potential as multi-targeted agents. However, further experimental validation is necessary to confirm these computational findings and evaluate the efficacy of these compounds in preclinical models. Pharmacokinetic and pharmacodynamic profiling revealed that most compounds exhibited favorable absorption, distribution, metabolism, and excretion (ADMET) properties, with 20 out of 22 compounds classified as highly absorbable and demonstrating good oral bioavailability. Notably, compounds such as C17 and C20 displayed the ability to cross the blood-brain barrier, suggesting potential utility in targeting breast cancer metastases to the central nervous system.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.











Comments on this article Comments (0)