Keywords
Aspergillus fumigatus, epithelial cells, bar-seq, infection model
This article is included in the NC3Rs gateway.
Aspergillus fumigatus, epithelial cells, bar-seq, infection model
Scientific benefit(s):
• Facilitates the identification of fungal genetic drivers of infection using a high throughput screening approach in mammalian cells.
• Can be easily adapted to screen different barcoded null mutant libraries for other pathogens, to understand mechanisms of infection in different cell types or to test drug efficacy during infection.
3Rs benefit(s):
• This method can be used to replace the use of animals in the screening of barcoded mutant strains.
• Represents an informed approach to select candidate gene deletion mutants for testing in animal models of disease thus facilitating a significant reduction for animal usage.
• Provides an ethically sustainable experimental framework to test multiple pathogen/cell line/drug combinations.
Practical benefit(s):
• Easily adoptable to carry out once barcoded mutant libraries are generated.
• High throughput, which can be further scaled up to screen thousands of mutants simultaneously.
Current applications:
Potential application(s):
Aspergillus fumigatus is the causative agent of aspergillosis and has recently been defined as a critical human fungal pathogen by the World Health Organisation.1,2 Aspergillosis is a leading cause of mortality and morbidity in immunosuppressed patients and those with a previous chronic respiratory condition. Treatment is restricted to four antifungal drug families, and drug resistance is becoming a global health problem.3 Therefore, a comprehensive understanding of the genetic armamentarium utilized by A. fumigatus to overcome antifungal host defenses is required to support the development of new treatments. In the lungs, A. fumigatus co-ordinately regulates multiple fungal adaptation and virulence mechanisms.4,5 This is the case for A. fumigatus protein kinases, which play crucial roles in various cellular processes including growth, stress response, and virulence.6–11 Until recently, our understanding of the A. fumigatus protein kinase complement was inferred from studies based on gene homology in A. nidulans.12 However, our group recently completed the annotation of protein kinases of A. fumigatus and generated a barcoded null mutant library consisting of 111 strains.13 Using this library, we demonstrated the utility of bar-seq in identifying genetic drivers of resistance to antifungal drugs and virulence in vivo.13
Bar-seq has emerged as a useful tool to assess microbial fitness under a range of conditions,14 and it has been pivotal in assigning new functions and growth phenotypes to genes of different bacterial species in single cultures,15 co-cultures,16 and in infection models.17 In yeast, bar-seq has also been used to elucidate the biological functions of genes and to understand stress responses and mechanisms of drug resistance.18–20 Here, we aimed to optimize a bar-seq protocol to understand which protein kinases are associated with survival within airway epithelial cells.
Airway epithelial cells have critical antifungal functions, as they can internalize and kill A. fumigatus spores within mature phagosomes.4,21–28 Activation of the antimicrobial responses of airway epithelial cells upon spore challenge also leads to the secretion of immune mediators, which attract professional phagocytes to further aid fungal clearance.25 The interplay between host and pathogen networks determines the outcome of the infection. A. fumigatus genes important for regulating conidial adhesion, internalization, and killing by airway epithelial cells have been defined by us and others (for review Ref. 29). To date, our understanding of Aspergillus factors associated with disease has been determined by assessing fungal infection in immunocompromised hosts using either neutropenic mice or mice with poorly targeted immunosuppression.30 These experiments are time consuming and costly, and their throughput performance is limited. Therefore, a high-throughput unbiased screening approach that allows the identification of A. fumigatus gene functions for the infection of airway epithelial cells, which can be adapted to other cellular systems, will enable the replacement of virulence studies that are currently based on a single mouse–one fungal strain model.
Here, we provide a comprehensive protocol for assigning new functions to A. fumigatus genes in driving airway epithelial cell infection. We have demonstrated the utility of this approach by screening our library of A. fumigatus deletion mutants in genes encoding protein kinases during infection of A549 alveolar epithelial cells. This epithelial cell line has been chosen because it is widely accepted to study the outcomes of fungal interactions with airway epithelial cells, as they produce results that correlate with clinically relevant phenotypes in whole animals and patients.4,22,31 Indeed, our group and others have exploited this cell line to identify critical events during the interaction of A. fumigatus with the host.4,21–23,25,26 The overall aim of this experiment was to identify A. fumigatus null mutants of protein kinases that are differentially associated with survival within A549 cells.
This protocol has been separated into four major sections, which are summarized in Figure 1. In section 1, we describe the culture and infection of A549 epithelial cell monolayers with a pool of barcoded A. fumigatus gene-knockout strains. In Section 2, we provide instructions for three different methods of removing extracellular A. fumigatus conidia, as well as comparative analysis, to allow the end-user to select the one that is most appropriate for them. These methods include pharmacological killing of A. fumigatus using nystatin,26 mechanical removal of extracellular conidia using cell surface washes23 and differential staining of intracellular and extracellular conidia coupled with fluorescence-activated cell sorting (FACS) of airway epithelial cells.21 Section 3 describes the extraction of genomic fungal DNA from A. fumigatus conidia within airway epithelial cells, and the preparation of a library for next-generation sequencing. Section 4 describes the bioinformatics pipeline used to analyze sequencing outputs, as well as the suggested validation experiments. Finally, we provide a dedicated section to validate the experiments to confirm the bar-seq observations.
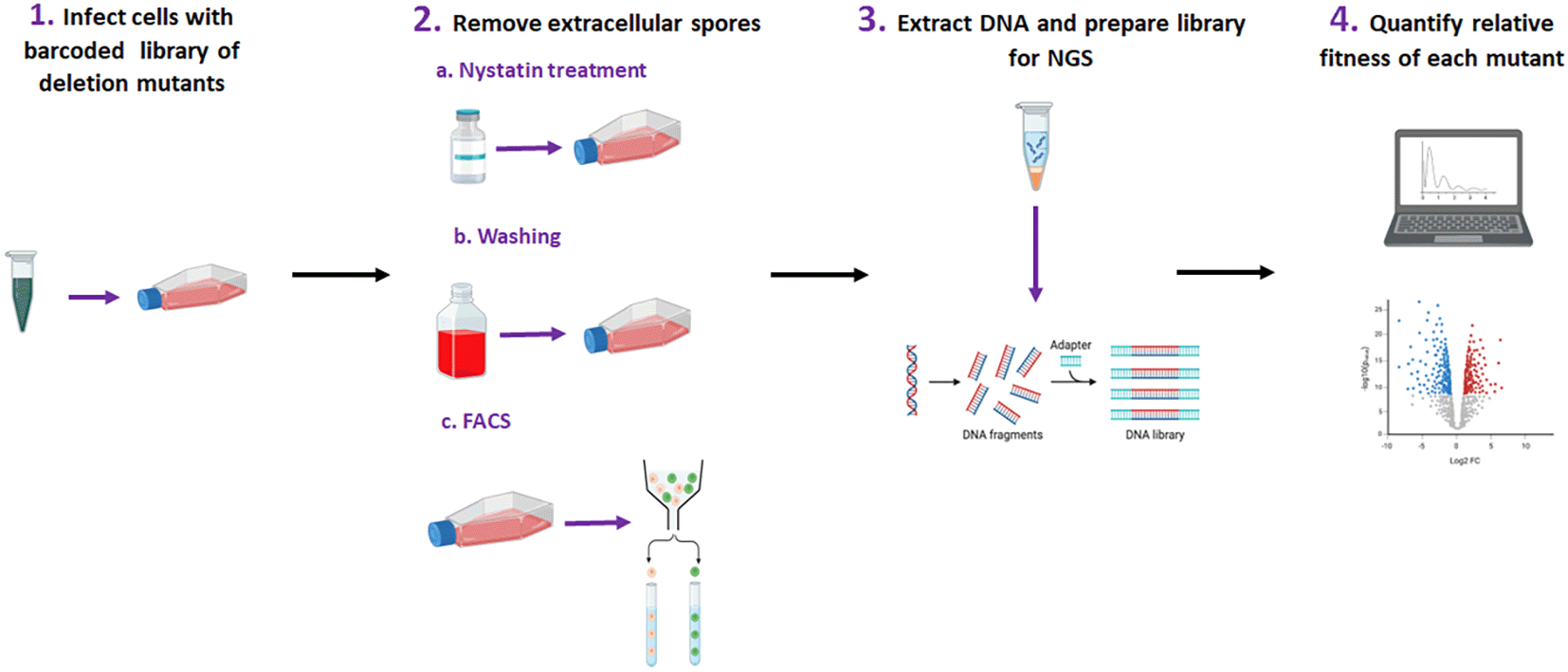
111 A. fumigatus protein kinase deletion mutants were obtained from the COFUN library. Strains were grown, and spores were harvested, counted and normalised to generate a pool of mutants. The pool was used to infect A549 airway epithelial cells at multiplicity of infection (MOI) of 1. The cells were then incubated for 6 h before the extracellular spores were removed via a) Nystatin treatment, b) Washing or c) FACS processing. These three experiments were carried out independently. Fresh media was then added, and the cells were incubated overnight to allow mycelia to grow. The mycelia were then harvested, DNA was extracted, and the barcoded region of each mutant was amplified via enrichment PCR. The library was prepared, samples were sequenced, and relative fitness was calculated using DESeq2 on raw counts. Results were normalised to the initial inoculum to keep consistency across methods.
A list of all the software, reagents, suppliers, and product codes can be found in Table 1.
Catalogue number is placed in brackets.
2.1.1 Preparation of the A. fumigatus library pool
The A. fumigatus kinase deletion mutant collection was previously generated using a high-throughput gene knockout method developed by our group, which combines fusion PCR to generate gene replacement cassettes with a multi-well transformation procedure. Our step-by-step protocol for the generation of A. fumigatus deletion mutants was comprehensively described by Zhao et al.32 and the composition of the full library, including the lists of primers used, was described by van Rhijn et al.13
1. Use sterile 10 μl loops to streak 111 A. fumigatus null mutant strains individually from glycerol stocks stored at -80°C. Strains were streaked onto 10 ml of Sabouraud Dextrose agar, poured into T25 cm2 flasks, and incubated for 3 days at 37°C.
2. Harvest conidia by adding ~10 ml of 0.01% phosphate buffered saline - Tween-20 (PBS-T) to T25 cm2 flasks and shaking gently to dislodge the conidia. Filter through Miracloth to remove mycelia and collect the conidia suspension in a falcon tube.
3. Quantify conidia using a Fuchs-Rosenthal haemocytometer
4. The concentration was normalized to 5 × 108 spores/ml and all strains were pooled into a single tube.
Note. Although A. fumigatus deletion mutant strains carry a hygromycin resistance cassette, we do not recommend growing the mutants in media with hygromycin before infection experiments, as this may induce stress that affects conidiation.
Note. It is critical that no mycelia are transferred to subsequent experiments, as this may create bias in bar-seq experiments. Therefore, we advise that conidia should not be dislodged from the media by scraping.
2.1.2 Seeding and infection of cells
1. Seed A549 (ATCC CCL-185) airway epithelial cells (7 × 105) in 5 ml of supplemented DMEM (sDMEM) in T25 cm2 flasks (we recommend 3-5 flasks per infection condition). sDMEM consisted of DMEM, 1% penicillin/streptomycin cocktail, and 10% fetal bovine serum (FBS).
2. Incubate the cells for 48 h at 37°C and 5% CO2 to achieve confluency.
Note. Before infection, using a light microscope check that there are not dead cells floating, that sings of vacuolisation in the cells are not visible and that monolayers are ~90% confluent. If enrichment of A. fumigatus conidia that has been internalized by A549 cells is performed by FACS, the A. fumigatus pool must be stained prior to infection of epithelial cells (described in section 2.2.3).
3. Remove old media from flasks using a sterile stripette and replace it with 5 ml of fresh sDMEM containing A. fumigatus spores adjusted to an MOI of 1.
a. Prepare A. fumigatus inoculum in 50 ml tubes and aliquote into T25 cm2 flasks containing confluent monolayers to reduce variation in the infectious dose between flasks.
Note: Confluent monolayers of A549 cells in T25 tissue culture flasks contain approximately 2.8×106 cells. Therefore, to achieve an MOI of 1, cells were infected with 2.8×106 spores.
4. Incubate at 37°C and 5% CO2 for 6 h to allow spore to be internalised.
To identify the most suitable method for removing extracellular conidia, we assessed three different approaches: 2.2.1) nystatin treatment of infected monolayers to kill extracellular A. fumigatus spores, 2.2.2) Washes of infected monolayers to remove unbound extracellular spores, and 2.2.3) differential fluorescence staining and fluorescence-activated cell sorting (FACS) of cells to select for internalized FITC-labelled A. fumigatus spores.
2.2.1 Nystatin treatment
1. Remove sDMEM from flask using a sterile stripette.
2. Wash cells gently three times with sDMEM pre-warmed at 37°C.
3. Nystatin (50 μg/ml final concentration) was added to 5 ml of sDMEM in flasks.33
4. Incubate cells for 1 h at 37°C, 5% CO2 to kill extracellular spores (note nystatin is expected to be excluded from mammalian cells; hence, internalized spores should remain viable).
5. Use a sterile stripette to remove sDMEM with nystatin, and to wash cells three times with PBS pre-warmed at 37°C to ensure drug is removed.
6. Then, add 5 ml of sDMEM and incubate the flask for 16 h at 37°C and 5% CO2 to allow intracellular A. fumigatus strains that have not been killed by A549 cells to grow.
2.2.2 Washing
1. Remove media from flasks and discard.
2. Add 5 ml of pre-warmed sDMEM to flasks, being careful not to disrupt cell monolayers. Gently swirl media around flask and remove it. Repeat this process for a total of 3 washes.
3. After final wash, remove sDMEM and replace with 5 ml of fresh sDMEM.
4. Incubate flasks for a further 16 h at 37°C and 5% CO2 to allow intracellular A. fumigatus strains that have not been killed by A549 cells to grow.
2.2.3 Fluorescence-activated cell sorting (FACS)
2.2.3.1 Fluorescein isothiocyanate (FITC) staining of pool (prior to infection)
1. The A. fumigatus library pool was centrifuged at 2,000 g for 3 min at room temperature , and the supernatant was discarded.
2. The pellet was then resuspended in 0.1 mg/ml FITC prepared in 0.1 M Na2CO3 and incubated in the dark for 30 min at 30°C.
3. Wash spores three times in 500 μl PBS, discarding supernatant each time.
4. The A. fumigatus library pool was resuspended in 1 ml PBS and counted using a hemocytometer.
2.2.3.2 Sample preparation for FACS (after 6 h of infection)
1. Label a 50 ml tube for each T25 cm2 flask.
2. Remove media from flasks and transfer to corresponding 50 ml tube.
3. The cells were gently rinsed with 5 ml of pre-warmed PBS and transferred to a 50 ml tube containing media.
4. Add 3 ml of Trypsin-EDTA to T25 cm2 flasks and incubate at 37°C and 5% CO2 for 5 min to detach the airway epithelial cell monolayer.
5. During this time prepare staining mix as described in 2.3.3, to identify extracellular spores.
6. Check the cells are fully detached using a light microscope before adding 3 ml of sDMEM to neutralize the trypsin-EDTA.
7. Transfer cell suspension to corresponding 50 ml tube.
8. Centrifuge the cell suspension at 520 × g for 3 min at room temperature before carefully pouring the supernatant.
9. The pellet was gently resuspended in a small volume of liquid and transferred to a sterile 1.5 ml tube.
10. Centrifuge cells at 240 g for 3 mins at room temperature and discard supernatant.
11. The cells were resuspended in 500 μl of the staining mix and incubated in the dark for 5 min.
12. Centrifuge samples at 240 g for 3 mins at room temperature and discard supernatant.
13. Resuspend cells in 500 μl of pre-warmed PBS and centrifuge again.
14. After the final centrifugation, the PBS was removed, and the cells were resuspended in 200 μl of pre-warmed PBS and kept on ice.
2.2.3.3 Preparation of staining mix
1. Dissolve 8 mg of Calcofluor White (CFW), which binds to chitin in fungal conidia, 1 ml of FluoroBrite media.
2. Dilute this stock 1:10 in FluoroBrite media to generate a 0.8 mg/ml stock.
3. In a 15 ml tube combine 500 μL Annexin V binding buffer with 5 μl of 0.8 mg/ml CFW per sample. A fresh working stock was prepared for each experiment and wrapped in foil to protect it from light.
2.2.3.4 FACS
1. A BD Influx Cell Sorter was used to sort epithelial cells that contained internalized A. fumigatus spores using a modified version of the gating strategy described in.22 Unlike22 the knockout library was constructed in a non-fluorescent background; therefore, A. fumigatus pooled mutants were pre-stained with FITC prior to infection. Epithelial cells that had internalized A. fumigatus were defined as FITC+/CFW-, whereas epithelial cells that had A. fumigatus attached to their surface were defined as FITC+/CFW+ and were excluded.
2. For each T25 cm2 flask, 10,000 epithelial cells containing A. fumigatus (stained FITC+/CFW-) were sorted into a 1.5 ml tube containing 500 μl of sterile H2O.
3. Vortex the 1.5 ml tubes containing the sorted samples to ensure that the epithelial cells are lysed and the internalized spores are freed. Transfer the 500 μl sample back into a T25 cm2 flask containing 5 ml of fresh sDMEM and incubate for a further 16 h at 37°C, 5% CO2.
2.3.1 Collection of mycelia
1. Collect fungal biomass into 15 ml tubes using a cell scraper.
2. Samples were centrifuged at 2,000 × g for 3 min at room temperature and as much liquid as possible was discarded.
3. Freeze overnight at -80°C.
4. Freeze dry samples to remove the remaining liquid (this allows multiple samples to be processed simultaneously, but a liquid nitrogen grinding method can also be used).
5. Grind frozen biomass into a fine powder using a pestle and mortar.
2.3.2 Extraction of fungal DNA
Note. In addition to extracting DNA from the A. fumigatus pools collected following infection, DNA should also be extracted directly from the pool of spores used to infect cells. This serves as a control to represent the actual composition of the pool.
1. The freeze-dried mycelia were resuspended in 1 ml of cetyltrimethylammonium bromide (CTAB) extraction buffer (2% CTAB, 20 mM EDTA·Na2·2H2O, 1.4 M NaCl, and 100 mM Tris, pH 8). When the liquid nitrogen grinding method was used, approximately 100 mg of ground mycelia was resuspended in 1 ml of CTAB buffer.
2. Transfer sample to a screw cap tube containing HCl washed glass beads.
3. The samples were bead-beaten using a Fastprep-24 instrument for 20 s at 4 s/m and incubated at 65°C for 10 min on a heat block. This step was repeated three times.
4. The sample was incubated at room temperature for 5 min before centrifuging for 5 min at 10,000 × g.
5. Transfer 700 μL of the supernatant to a sterile 2 ml tube and add 4 μL of RNase A (100 mg/ml).
6. Incubate sample at 37°C for 20 min.
7. Then, 700 μL of chloroform:isoamyl alcohol (24:1) was added to the sample and vortexed for 10 s.
8. The sample was centrifuged for 2 min at 10,000 × g, and ~600 μL of the aqueous phase was transferred to a sterile 2 ml tube. The transferred sample should be clear and this step should be repeated if it remains cloudy.
9. Then, 360 μL of isopropanol was added to the sample and inverted a few times to mix. The samples were kept on ice from this point onwards.
10. Centrifuge for 10 min at 10,000 g at 4°C, and discard supernatant. This should be performed carefully to avoid dislodging pellets.
11. The pellet was washed twice with 500 μL 70% ethanol and allowed to air-dry at room temperature.
12. Once the ethanol had fully evaporated, the DNA was resuspended in 20 μL of molecular grade nuclease-free H2O.
2.3.3 Sequencing preparation
A) Enrichment of barcode region in deletion mutants
1. Amplify the barcode region of mutants by PCR ( Table 2) using universal primers that include Nextera compatible adapters (underlined):
PCREnrichFw5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCGGCTCGGTAACAGAACTA-3′
PCREnrichRv 5′- GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGTCGTTGTAGGGGCTGTAT-3′
2. Check PCR product (264 bp) using 1% agarose gel.
B) Clean-up of PCR product using AMPure XP beads
1. Transfer 18 μl of PCR product to a 1.5 ml tube containing 0.8X AMPure beads and mix gently by pipetting up and down ten times. This process removes fragments of < 200 bp.
2. Incubate samples at room temperature for 5 min.
3. Place tubes on a magnetic stand for 2 min to allow beads to separate.
4. Discard supernatants and wash beads twice with freshly prepared 80% ethanol.
5. Air-dry the beads for 10 min, remove the tubes from the magnetic stand, and add 36 μl of nuclease-free water. The beads were checked periodically to prevent overdrying.
6. Gently pipette up and down ten times to resuspend the beads fully.
7. The samples were incubated at room temperature for 2 min and then returned to a magnetic stand for 2 min.
8. The beads were allowed to separate and 30 μL of the eluates was transferred to new tubes. samples can be kept at -20°C until further use.
C) Next-generation sequencing library preparation
1. The amplicon library is prepared using Nextera XT v2 DNA Library Prep Kit according to the manufacturer’s protocol summarized in Table 3.
Note. For iSeq100, it is necessary to ensure that at least one index does not start with two G bases; otherwise, it cannot be called effectively. See the Illumina guide to determine which Nextera XT Index Primer combinations are suitable for your samples.
2. Check PCR product (280 bp) using 1% agarose gel.
D) Second clean-up of PCR product using AMPure XP beads
1. Transfer 30 μl of PCR product to a tube containing 33 μl of AMPure XP beads and mix gently by pipetting up and down ten times.
2. Incubate samples at room temperature for 5 min.
3. Place tubes on a magnetic stand for 2 min to allow beads to separate.
4. Remove supernatants and wash beads twice with freshly prepared 80% ethanol.
5. Air-dry the beads for 10 min, remove the tubes from the magnetic stand, and add 36 μl of nuclease-free water. The beads were checked periodically to prevent overdrying.
6. Gently pipette up and down ten times to resuspend the beads fully.
7. The samples were incubated at room temperature for 2 min and then returned to a magnetic stand for 2 min.
8. The beads were allowed to separate and 25 μL of the eluates was transferred to new tubes. samples can be kept at -20°C until further use.
E) Library quantification and sequencing
Note. Because the A. fumigatus knockout kinome library contains only 111 individual mutants, samples can be run using an Illumina iSeq-100 platform. For larger libraries, platforms with increased sequencing capacity should be considered.
1. Quantify samples using Qubit dsDNA HS-Assay-Kit following manufacturer’s instructions.
2. Calculate molar concentration of DNA based on amplicon size after index PCR reaction.
3. Dilute samples to 1 nM in 10 mM Tris pH 8.5.
4. Add 5 μl of each sample together in a single tube.
5. Dilute pooled samples from 1 nM to 50 pM in 10 mM Tris pH 8.5.
6. Spike samples with 5-20% PhiX Control library to increase the diversity of the sequencing pool.
7. Load 20 μL of 50 pM libraries with PhiX control into an iSeq-100 reagent cartridge (Illumina) and sequence on an iSeq-100 platform to generate 2 × 150 bp paired-end reads.
Final concentration for primers is shown in brackets.
| Reagent | Volume |
|---|---|
| 2X Phusion Master Mix | 20 μl |
| Forward/Reverse Primer | 2 μl (5 μM) |
| DNA | 4 μl |
| PCR water | 22 μl |
| PCR conditions | ||
|---|---|---|
| Initial step | 98°C | 30 s |
| 35 cycles | 98°C | 5 s |
| 55°C | 30 s | |
| 72°C | 1 min | |
| Final step | 72°C | 10 min |
| Reagent | Volume |
|---|---|
| 2X KAPA HiFi HotStart ReadyMix | 25 μl |
| Nextera XT Index Primer 1 (N7xx)/Nextera XT Index Primer 2 (S5xx) | 5 μl |
| Cleaned PCR product (obtained in 3.3B) | 5 μl |
| PCR grade water | 10 μl |
| PCR conditions | ||
|---|---|---|
| Initial step | 95°C | 3 mins |
| 8 cycles | 95°C | 30 s |
| 55°C | 30 s | |
| 72°C | 30 s | |
| Final step | 72°C | mins |
2.4.1 Computational analysis
Note: All codes for the analysis and plots are available in Ref. 34.
1. FASTQ files were obtained from the BaseSpace Sequence Hub (Illumina) and checked for quality using FastQC. Poor quality reads were discarded.
2. Raw sequencing reads were trimmed using CUDADAPT (v.4.9) which removes the adapter sequence GGTAACAGAACTA from the 3′ end of the reads and discard reads shorter than 21 bp and longer than 23 bp.
3. Generate an index file with the AFUB number and a unique barcode sequence for each mutant strain. Bowtie2 (version 2.5.0) was used to map the reads to this index.
4. SAM tools (v.1.2.) was utilised to convert and index SAM files to BAM format and obtain counts using the Perl script.
5. DESeq2 (v. 1.38.3) was used to calculate the differential barcode counts for each mutant on a condition versus standard basis (default settings). A comprehensive explanation of this can be found in Ref. 35.
6. Log-transform normalized counts were produced by DESeq2 and plotted using pheatmap (v. 1.0.12) or GraphPad Prism v10 (La Joya, CA, USA) ( Figure 2).
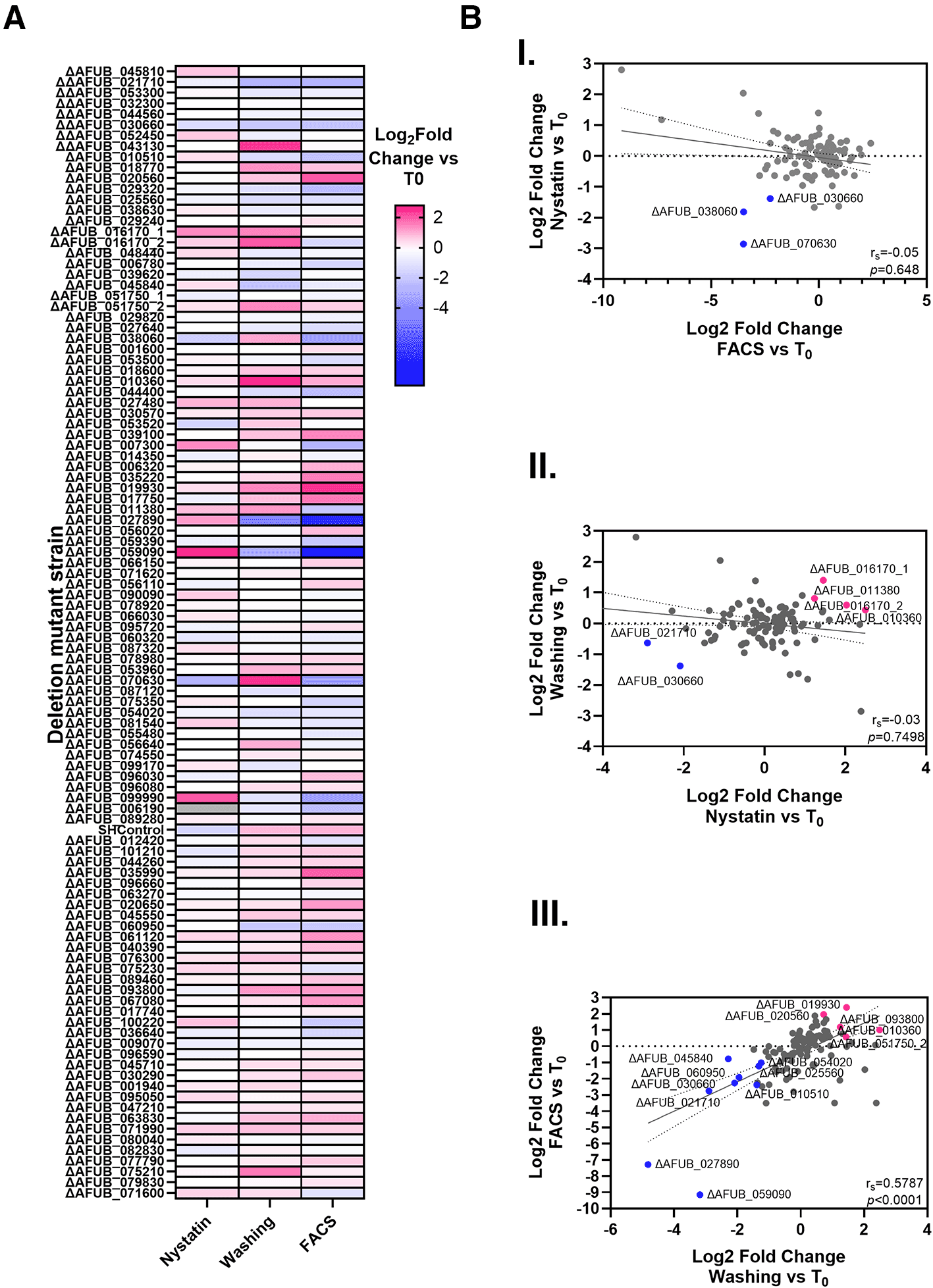
(A) Heatmap of fitness for each A. fumigatus protein kinase null mutant relative to T0 . Relative fitness values was calculated by DESeq2, comparing barcode counts obtained from A549 cells infected with the pool for 16 hours following removal of extracellular conidia by Nystatin treatment, PBS washes or FACS sorting. 103 individual mutants were detected. (B) Global correlation analyses of population dynamics of A. fumigatus deletion mutant libraries during infection of A549 cells and removal of extracellular conidia by nystatin treatment, washing the epithelial monolayer or sorting the population of cell containing internalised spores. Spearman correlation index (rs) and P values are shown for each comparison.
2.4.2 Validation of observations
Having identified those A. fumigatus individual mutants that are differentially abundant, carry out validation of the results utilizing killing experiments.23
i. Seed 7.5×104 A549 cells in 500 microlitres of sDMEM and incubate for 48 h at 37°C and 5% CO2. At this point, cells were 90% confluent.
ii. Before infection, the media was changed, and the cells were exposed to 50 μL of A. fumigatus spores containing 106 spores.
iii. Incubate infected cells for 4 h at 37°C, 5% CO2, wash extracellular spores with pre-warmed PBS, and add 500 μL of sDMEM. Infected cells were incubated for an additional 12 h.
iv. Serial dilutions of the cell wash were made and plated on Sabouraud Agar plates to calculate spore input. Incubate the plates for 24 h at 37°C.
v. After the cell incubation, the medium was removed, 500 μL of sterile deionized water was added, and serial dilutions were made. 50 μL of each serial dilution were plated on Sabouraud Agar plates and incubated for 24 h at 37°C.
vi. Spore survival for each individual mutant and parental strain was calculated. Experiments were carried out in biological and technical triplicates.
Note: Growth rate analyses of individual mutants in culture media are recommended to ensure that the observed phenotypes are not a result of mutant growth defects.
• Spearman correlation tests were performed to determine the consistency across methods. Analyses were performed using GraphPad Prism v10 (La Jolla, CA, USA).
• Statistical differences in abundance for each mutant were determined using Bowtie 2 as described above.
• One-way ANOVA with Geisser-Greenhouse (repetitive measurements) and Dunnett’s correction (multiple comparisons) were performed to determine differences in fungal survival and growth rates between mutants and controls.
• P<0.05 or Padj<0.05 were considered for statistical significance.
This method is based on the use of the well-characterized A549 alveolar epithelial cell line, as it provides highly reproducible outputs from A. fumigatus-host interactions, which exhibit a high degree of correlation with studies using primary airway epithelial cells and mice. All experiments were conducted with 3-5 technical replicates for each of the 3-5 biological replicates. P-values for test treatments versus controls were calculated through parametric or non-parametric tests according to normality distribution. p<0.05 was utilized in all cases as a threshold for statistical significance. Statistical significance for differential abundance of the null strains was analyzed using Deseq2, as explained above, and a Padj<0.05 is utilised as a statistically significant cutoff.
Airway epithelial cells are the first point of contact for inhaled A. fumigatus spores with the host; however, our understanding of the fungal drivers of A. fumigatus infection of these cells remains limited. We have previously shown that a barcoded A. fumigatus gene knockout kinase library can be competitively analyzed to identify new genetic drivers of antimicrobial resistance and virulence in vitro and in whole animals.13 Thus, we performed in vitro competition experiments in the presence of an immortalized epithelial cell line, A549, to investigate the initial steps of A. fumigatus colonization of the lung.
To identify individual kinase deletion mutants that are differentially abundant during infection with airway epithelial cells, we utilized three different approaches to remove extracellular A. fumigatus: (i) nystatin treatment, (ii) washing, or (iii) FACS sorting of airway epithelial cells containing A. fumigatus conidia ( Figure 1). After infection, fungal DNA was extracted and the ratio of absorbance at 260 nm and 280 nm was confirmed to range from 1.8 to 2 for all samples; DNA was also size separated using gel electrophoresis to confirm integrity. DNA gel electrophoresis after enrichment PCR confirmed amplification of the 280 bp barcode PCR fragment. Gel electrophoresis was performed after indexing and clean-up steps before loading the sample into the sequencer to visually determine the presence of the DNA library (Supplementary Figure 1,34).
The expectation was that all three experiments, which were designed to assess the impact of gene knock-out on a combination of cellular uptake, spore killing, and host fitness, would generate similar outputs; however, this was not the case. Although a reasonable correlation was observed between FACS sorting and spore washing approaches (rs=0.5787), and this correlation was statistically significant (p < 0.0001), no correlation was observed between these approaches and nystatin treatment ( Figure 2). This may indicate, contrary to our expectation, that nystatin treatment is not effective at differentiating internalized from external spores.
A total of nine null mutants were identified as differentially abundant in the data generated from the washing approach (four increased abundance, five decreased abundance), and 15 mutants were differentially abundant in the FACS data (2 increased, 13 decreased) ( Figure 2C and supplementary Figure 2,34). To validate the results of these experiments, we selected two A. fumigatus deletion mutants that were either more (ΔAFUB_020560, null in the rck2 ortholog of Saccharomyces cerevisiae36) or less abundant (ΔAFUB_059090 null in the nrc2 ortholog of Neurospora crassa37) in both datasets ( Figure 2B). Having confirmed that these mutant strains did not show differences in growth compared to the parental strain when grown in culture media ( Figure 3A), we performed killing assays to assess their susceptibility to exposure to airway epithelial cells ( Figure 3B). Our data revealed that, consistent with the bar-seq data, the ΔAFUB_020560 mutant was more able to survive within airway epithelial cells than the parental strain, and that the ΔAFUB_059090 mutant was more susceptible to airway epithelial cell killing.
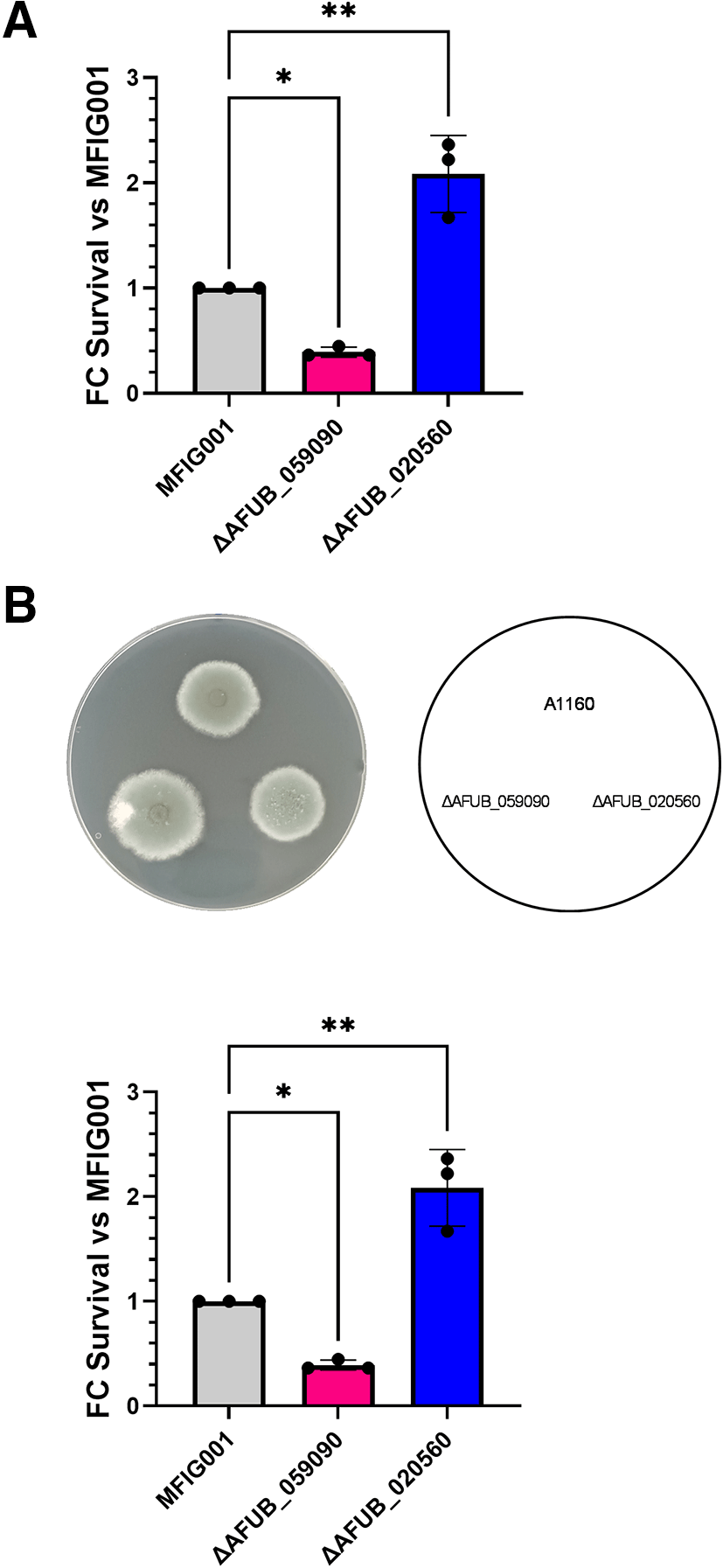
(A) A. fumigatus survival of individual null mutant strains identified by bar-seq analyses during infection of A549 airway epithelial cells (n = 3). (B) Growth rate analyses of individual A. fumigatus null mutant strains. * p < 0.05, p < 0.01.
Together, our data indicate that analysis of barcoded A. fumigatus deletion libraries allows the identification of the drivers of fungal survival within airway epithelial cells. Our data also show that removal of extracellular conidia via iterative washing or FACS-mediated sorting of airway epithelial cell populations that have internalized A. fumigatus is superior to pharmacological killing of extracellular conidia with nystatin. We recommend the use of FACS-mediated sorting whenever possible as it decreases the likelihood of enriching strains that might not be fully internalized.
To date, much of our understanding of A. fumigatus pathogenicity has been derived from targeted studies using individual knockout mutants in infection experiments. Although effective, this process can be laborious and time-consuming. In contrast, the emergence of bar-seq technologies represents a powerful tool for high-throughput screening of deletion mutants, enabling us to simultaneously ascertain their functions under a variety of different conditions. This technology has been used extensively in both bacteria and other fungal species, primarily to assign functions to uncharacterized genes, investigate tolerance to antimicrobials, and identify targets for novel therapeutics.15,19,20 Most recently, functional screens have been carried out in model organisms such as Saccharomyces cerevisiae, Candida albicans, and A. fumigatus to uncover genotypes involved in conferring fitness both in vitro and in vivo.13,38 Phadke et al.38 carried out a genome-wide screening of S. cerevisiae to identify genes contributing to pathogenicity in a Galleria mellonella infection model, whereas van Rhijn et al.13 investigated the A. fumigatus kinome to identify genes that promote hypersensitivity to azoles and fitness defects in a murine host. However, the applicability of bar-seq to investigate the genetic armamentarium A. fumigatus that can survive within host immune cells has not been explored.
In our study, we optimized an experimental approach to screen barcoded mutant libraries during infection of airway epithelial cells. We comparatively analyzed the performance of three methods commonly used to remove extracellular conidia in A. fumigatus infection experiments (see Methods section 2). Our data suggest that nystatin treatment might not completely remove extracellular A. fumigatus, thus failing to accurately resolve A. fumigatus protein kinases and facilitating fungal survival within airway epithelial cells. This might result from the use of nystatin at fungistatic rather than fungicidal concentrations to avoid toxicity to host cells.33 In contrast, we found a significant positive correlation for the identification of A. fumigatus kinase genes contributing to fitness during in vitro infection when extracellular conidia were removed either by washing or FACS. Indeed, in addition to the identification of mutants that could be internalized and persist, washing of the cell monolayer also enriched the results with deletion strains, which might have an increased ability to adhere to the cell, which is an important step in colonization. Conversely, FACS included only deletion strains that were internalized by epithelial cells, producing a cleaner set of results. Extended data34 showing the mean number of reads for each mutant (see Earle F1000 NC3Rs Gateway Raw Data, Supplementary Figures 2A-C tabs) supports this theory, with a much larger number of reads obtained for the washing condition compared to FACS. Nevertheless, validation experiments specifically quantifying the adhesion of the mutants are necessary to confirm this. However, the use of FACS is limited to locations where this technology is available. We have also uncovered important factors to consider during experimental design, including ensuring sufficient cells are sorted during FACS to ensure that each mutant is sufficiently represented in the final dataset and to prevent a lack of experimental power due to the low number of barcode sequencing reads. We recommend that when using a sorting approach, at least 100 host cells per A. fumigatus deletion mutant are sorted to have sufficient sequence coverage per mutant strain. However, a titration experiment may need to be carried out when using barcoded mutant libraries with an increased number of strains. To conclusively identify the role of the discovery factors in fitness during infection of airway epithelial cells, a validation experiment must be designed. This experiment needs to be focused on confirming (i) that the mutants identified are not differentially fit to grow in the presence of airway epithelial cells due to growth defects, and (ii) that the identified mutants retain their fitness phenotype when analyzed individually in infection experiments. For the first point, users might be interested in including a control to analyze the fitness of the pool in culture media alone. For the purpose of this paper, we chose to compare with the initial mutant pool alone, as our aim was to perform correlation analysis across the three different methodologies, which provided a consistent starting point. Moreover, the library utilized in these experiments has already been extensively characterized for growth defects in previously published studies.13 Second, we have assigned new roles to two A. fumigatus protein kinases in fitness during infection of airway epithelial cells, providing a strong foundation for further investigations on these targets identified in our screens.
In Saccharomyces cerevisiae the rck2 gene36 regulates oxidative and osmotic stress; however, whether this is the case in A. fumigatus should be determined. The A. fumigatus gene AFUB_059090 encodes a protein kinase that is the nrc2 ortholog of Saccharomyces cerevisiae. In yeast, null mutants of this gene produce conidia that do not mature normally and hyphae that are thinner.37 Although we have not explored these phenotypes in detail in A. fumigatus it might be the case that these morphological changes facilitate spore killing by airway epithelial cells, resulting in fitness deficiency.
From a 3Rs perspective, it is crucial to emphasize that the use of this high-throughput technology in our cell culture system will allow the complete replacement of the use of animals for screening purposes, while enabling a significant reduction in the number of fungal strains that need to be tested in animals for full characterization. For example, assessing the individual role of virulence of the 111 A. fumigatus mutant strains in the kinome library using an immunosuppressed mouse model of invasive aspergillosis would require the use of 1221 mice (111 groups of 11 mice). However, the same approach using in vitro competitive fitness would require the use of only 10 T-25 tissue culture flasks and will allow us to triage those strains to be studied in an animal model, potentially leading to a 97% reduction in mice used (three groups of 11 mice).
By coupling the use of this methodology with isogenic cell systems deficient in particular host antifungal gene functions (e.g., ZNF77,24 p1131) we will also be able to identify the mechanisms of fungal infection in genetically susceptible populations. Similarly, this methodology could be applied to understand the specific mechanisms of fungal pathogenicity using primary cells from aspergillosis in at-risk patient cohorts (e.g., those with chronic respiratory conditions or cystic fibrosis).
Our group has pioneered the development of the A. fumigatus whole-genome knockout library (COFUN), which consists of more than 6,000 barcoded deletion mutants in individual genes. Evaluation of virulence defects for each mutant in this collection would require the use of approximately 66,000 mice, using existing technologies. However, the same approach using in vitro competitive fitness would require the use of a handful of tissue culture flasks and would allow us to triage those strains to be studied in an animal model. For example, if 10 mutant strains are identified in in vitro screening, assessing those strains for virulence in an animal model would require only 110 mice (85% reduction).
Overall, this study presents an optimized method for high-throughput screening of barcoded A. fumigatus mutant libraries to identify novel fungal genes that confer fitness in mammalian cells. The methodology proposed here is very versatile, and the current efforts for the generation of the A. fumigatus whole genome knockout library (COFUN library) offer new opportunities for genome-wide analyses of A. fumigatus drivers of infection with mammalian cells. Combining bar-seq and functional validation experiments, we will be able to streamline the identification of A. fumigatus deletion mutants involved in pathogenesis and reduce the number of strains to be tested in in vivo models of disease.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)