Keywords
Whole Genome Sequencing, SARS-CoV-2, Vaccine Candidate
This article is included in the Nanopore Analysis gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the Coronavirus (COVID-19) collection.
Whole Genome Sequencing, SARS-CoV-2, Vaccine Candidate
The COVID-19 pandemic in East Java, Indonesia has profoundly affected the region, resulting in a substantial number of cases and elevated mortality rates.1 The first confirmed case of COVID-19 in East Java was documented on March 11, 2020, involving a 50-year-old woman who had recently returned from Saudi Arabia and tested positive for the virus.2 Since that time, the incidence of cases has consistently risen, resulting in pressure on the hospital system and public health infrastructure.3 The COVID-19 pandemic, instigated by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has necessitated the urgent discovery of effective vaccines to control the virus’s transmission and alleviate its effects on public health.4 Indonesia has previously succeeded in developing an inactivated platform SARS-CoV-2 vaccine that has been clinically tested and widely used for the Indonesian population.5–9 However, there are concerns about the emergence of various new variants that could reduce the efficacy of the current vaccines, necessitating the development of a new vaccine with an mRNA cocktail platform derived from various SARS-CoV-2 variants, which is expected to help combat future COVID-19 outbreaks. Exploration of potential isolates to be developed as vaccine candidates is a crucial and important initial step.
Whole genome sequencing and phylogenetic analysis of SARS-CoV-2 isolates have become essential methodologies for elucidating the genetic diversity, evolution, and prospective vaccine targets of the virus.10–12 Phylogenetic analysis enhances whole genome sequencing by examining the evolutionary links among various viral strains.13 Researchers can reconstruct the phylogenetic tree by comparing the genetic sequences of SARS-CoV-2 isolates from East Java with those from other places, yielding information about the virus’s genesis, dissemination, and transmission patterns. This information is essential for comprehending the virus’s evolution and dissemination throughout the local population.14,15 Additionally, whole genome sequencing and phylogenetic analysis can identify conserved sections of the viral genome that are less prone to mutation and may serve as targets for vaccine development. These areas may encompass highly immunogenic epitopes or critical viral proteins implicated in viral replication and host interactions. By focusing on these conserved areas, novel platform vaccines can offer extensive protection against several viral types.16
This study aims to conduct whole genome sequencing, phylogenetic analysis, and mutational assessment of SARS-CoV-2 potential isolates from East Java, Indonesia, to find possible next-generation vaccine candidates. The results will enhance comprehension of the genetic variety, evolution, and prospective vaccine targets of the virus in this area. This research is essential for directing the creation of innovative and efficacious vaccines against SARS-CoV-2, especially in regions significantly affected by the disease, such as East Java. Utilizing whole genome sequencing and phylogenetic analysis, we can facilitate the creation of novel vaccine platforms capable of managing the COVID-19 pandemic and future outbreaks of related coronaviruses.
This research used isolates from Research Center for Vaccine Technology and Development stored at -80oC. The isolates were clinical samples of nasopharyngeal and oropharyngeal swab obtained from patients with COVID-19 positives on the rapid antigen test.
Virus isolation and propagation commenced with the inoculation of a Vero Cell Line (CCL81, ATCC) obtained from cell bank of Research Center for Vaccine Technology and Development, Universitas Airlangga, Indonesia, into a T25 flask (NEST®, China), prompted by the detected low viral concentration in the inoculum samples. One milliliter of sample was introduced into a T25 flask containing 80% confluent Vero CCL-81 cells after the removal of the cell culture medium, followed by incubation at 37°C with 5% CO2 for one hour, with gentle agitation every 15 minutes. A total of 7 ml of Minimum Essential Medium (Gibco, USA, Cat.No. 61100-061) supplemented with 5% Fetal Bovine Serum (Gibco, USA, Cat.No. 10437-028), 1% Penicillin-Streptomycin (Gibco, USA, Cat.No. 15140-122), and 1% Amphotericin B (Gibco, USA, Cat.No. 15290-026), was placed into a T25 flask and incubated at 37°C with 5% CO2 for 24 hours. The presence of cytopathic effect (CPE) was monitored daily using an inverted microscope. The CPE was monitored and subsequently transferred to the T75 flask. To enhance replication, virus isolates were put to a T300 flask containing Vero E6 cells. Virus isolation and propagation were validated through CPE assay and RT-PCR (The Applied Biosystem™ QuantStudio ™ 5 Real-Time PCR System, Thermo Fisher Scientific, USA). Isolates exhibiting elevated TCID50 (50% Tissue Culture Infectious Dose) values in the CPE assay were utilized for further characterization.
The chosen isolates underwent viral nucleic acid extraction with the ReliaPrepTM Viral Total Nucleic Acid Purification Kit (Promega). The RNA was measured using the Qubit RNA HS test kit and the Qubit 4.0 fluorometer. RT-qPCR was conducted utilizing the AllplexTM 2019-nCoV Assay, focusing on the RNA-dependent RNA polymerase (RdRp) gene, E gene, and N gene. A total of 17 μL of the mastermix was generated by including 5 μL of 2019-nCoV MOM, 5 μL of 5X Real-time, 5 μL of One-step Buffer, 2 μL of Real-time One-step Enzyme, and 5 μL of RNase-free Water. An appropriate internal control was employed for each PCR run. Eight microliters of each RNA sample were added to the tube containing the One-step RT-PCR Mastermix. 2019-nCoV served as the positive control, whereas RNase-free water functioned as the negative control. The sequences of the primer and probe were maintained in confidentiality. The thermal cycling technique included reverse transcription at 50°C for 20 minutes, followed by reverse transcription inactivation at 95°C for 15 minutes, and thereafter 45 cycles of 94°C for 15 seconds and 58°C for 1 minute.
Nine SARS-CoV-2 samples with strong positivity and stability were evaluated for whole genome sequencing utilizing Oxford Nanopore Technologies. An ideal CT value is necessary according to the ARTIC network nCoV 2019 sequencing methodology v3 LoCost, based on the CT value from the RT-qPCR test. If the CT value ranges from 12 to 15, a 100-fold dilution in nuclease-free water is required; if the CT value is between 15 and 18, a 10-fold dilution is necessary; and if the CT value exceeds 18, the sample should be processed without dilution. cDNA was generated from 8 μL of viral RNA utilizing LunaScript RT SuperMix reagents and primers. PCR was conducted utilizing Q5 Hot Start High-Fidelity 2X Master Mix and NEBNext ARTIC SARS-CoV-2 Primer Mix. The PCR mixture was first incubated for 30 seconds at 98°C for initial denaturation, followed by 35 cycles of 95°C for 15 seconds and 63°C for 5 minutes. The amplified products were purified using NEBNext Sample Purification Bead. The purified DNA was processed using NEBNext Ultra II End Prep mastermix (New England Biolabs, USA), followed by barcoding using EXP-NBD104/114 kit (Oxford Nanopore Technologies, UK) to obtain the DNA ends. The final concentration of DNA was measured using dsDNA HS Assay Kit (Thermo Fisher, USA) and calculated using Qubit™ 4 Fluorometer (Invitrogen™, Thermo Fisher Scientific, USA). Priming the flow was conducted before inserting the sample into the flow cell. The final DNA sample of 60 ng was added to the DNA library with a final volume of 75 μL (Oxford Nanopore Technologies, UK), followed by the ligation process using NEBNext Quick Ligation Mastermix (New England Biolabs, USA). Whole genome sequencing was performed on the flow cells using the Minion MinKnow software version 24.06.16 (Oxford Nanopore Technologies, UK).
The Multiple Sequence Comparison by log-Expectation (MUSCLE) was utilized to conduct sequence alignment, accessible at the following website: https://www.ebi.ac.uk/jdispatcher/msa/muscle. The phylogenetic tree was constructed utilizing the maximum likelihood method through Molecular Evolutionary Genetics Analysis across Computing Platforms (MEGA XI). The tree topology underwent assessment through one thousand bootstrap replicates. The complete genome sequences of the nine chosen isolates were examined to detect mutations in the protein-coding regions and to compare these sequences with the reference SARS-CoV-2 genome (hCoV-19/Wuhan-Hu-1/2019) obtained from the Global Initiative for Sharing All Influenza Data (GISAID, https://www.gisaid.org/) database. The viral genetic sequence and mutations were analyzed using the Nexclade online platform (https://clades.nextstrain.org), with comparisons made against the wild-type Wuhan-Hu-1 (NC_045512.2).
The alpha variant was initially identified in Indonesia in January 2021, with limited reports concerning its presence in the Indonesian population.17 The first case of the Delta variant of SARS-CoV-2 was detected in June 2021, resulting from a screening performed during the Suramadu lockdown. Three people tested positive for both antigen and PCR, exhibiting a low CT value.18 The Delta variety, previously designated as lineage B1617.2, originated in India. The Delta variant significantly influenced the dramatic increase in cases and deaths recorded from June to August 2021. The Omicron variant of SARS-CoV-2 was first identified in East Java in January 2022, resulting in a substantial rise in patient numbers until March. From January to March 2022, there was no notable rise in the mortality rate ( Figure 1).19
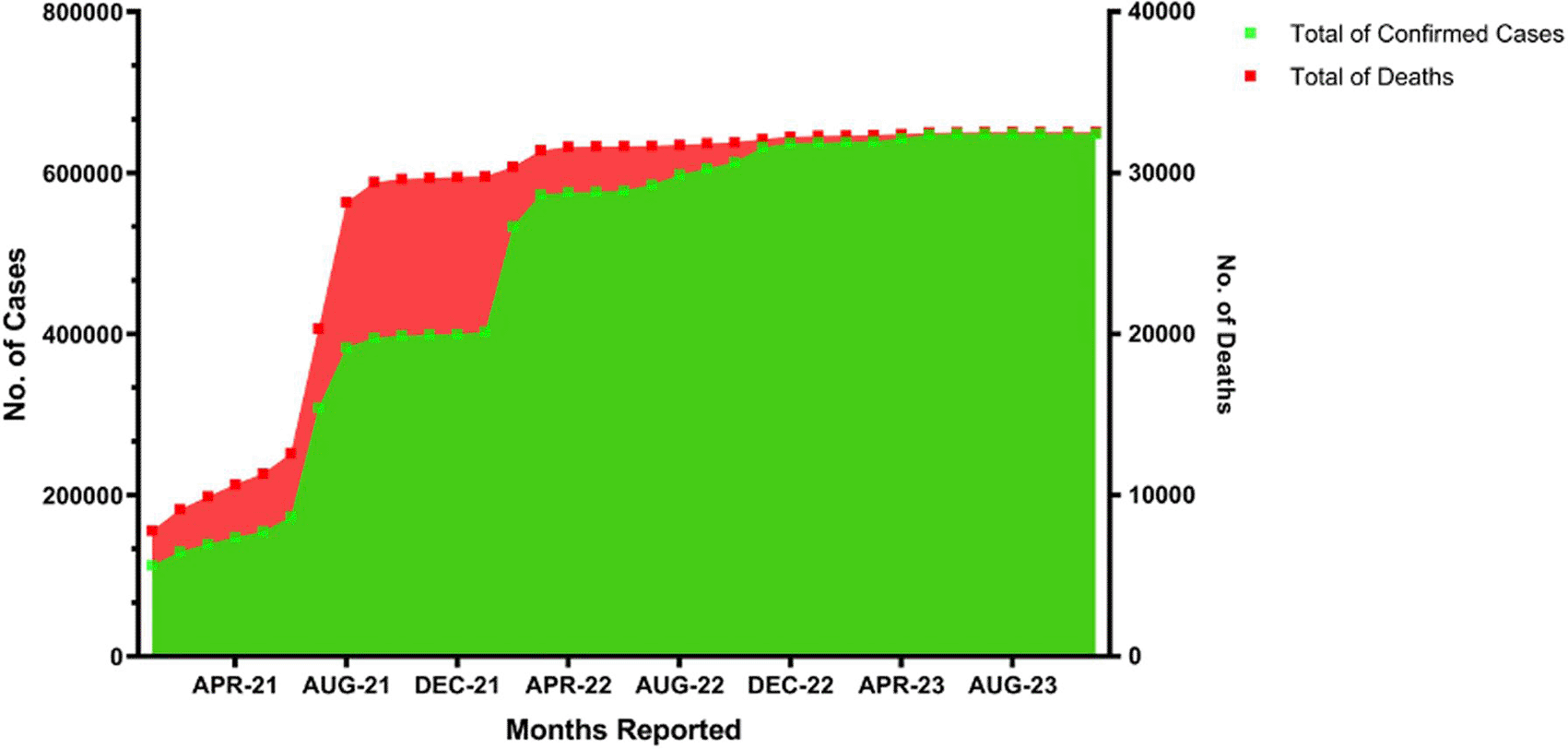
The data were obtained from Government of Indonesia (https://covid19.go.id/peta-sebaran ).
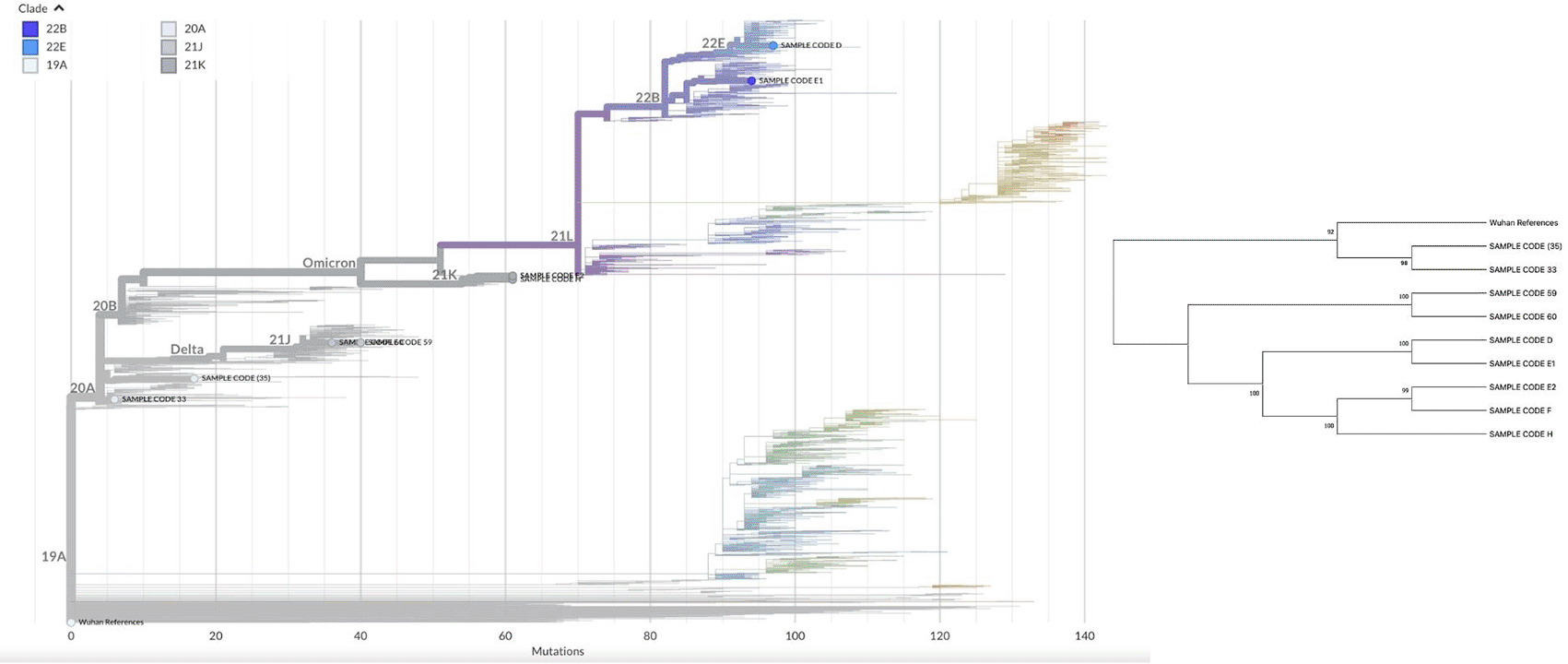
The selection of appropriate isolates was based on their performance in isolation and propagation on CCL81 and E6 Vero cells, exhibiting vigorous virus growth, plaque formation, and elevated virus titers. Nine isolates were obtained, indicating potential for subsequent study, including molecular characterisation. MINIon nanopore sequencing was utilized to perform sequencing tests. Sequencing analysis of 9 isolates indicated that 4 samples were identified as the 2 samples of Alpha and 2 samples of Delta variant of the SARS-CoV-2 virus, whereas 5 samples were identified as the Omicron variant ( Table 1). The samples identified as the Alpha and Delta variant belong to a singular lineage, B.1 and AY.23, respectively, whereas the Omicron variant exhibits several lineages, including three samples of lineage BA.1.1, one sample of BA.5.2, and one sample of BQ.1.23. Phylogenetic analysis of the whole genomes revealed five distinct clades: 20A (33,35), 21J (59, 60), 21K (E2, F, H), 22B (E1), and 22E (D) ( Figure 2).
Genetic mutations were identified in nine selected isolates. The nine isolates displayed differences in the number of mutations seen in nucleotides and amino acids ( Figure 3). The frequency of nucleotide mutations exhibits a positive correlation with the temporal appearance of variants, unlike the frequency of amino acid mutations. The predominant nucleotide and amino acid modifications were seen in the spike structural protein, as opposed to the other structural proteins. The spike protein enables target identification, cellular entry, and ultimately the viral infection that leads to varying levels of COVID-19 severity. The quantification of SARS-CoV-2 amino acid mutations revealed that the average mutation rate for the spike protein is around 46.96%, followed by ORF1a at 15.59%, ORF1b at 15.10%, N at 7.04%, M at 5.32%, ORF7a at 1.97%, ORF9b at 1.85%, ORF3a at 2.71%, E at 2,40%, ORF7b at 0,79%, and ORF6 at 0.26% ( Figure 4).


The Delta variant evolved from a variant under investigation (VUI) to a variant of concern (VOC) upon its classification within the B.1.617.2 lineage. The reclassification was predicated on an assessment of its transmissibility, which was demonstrated to be at least comparable to B.1.1.7 (Alpha variant). The mutation is thought to be partially responsible for the commencement of India’s deadly second wave of the pandemic, which began in February 2021. By the end of July, it had also caused an increase in daily infections across various parts of Asia,20 the United States,21 Australia, and New Zealand. During the Delta epidemic, the initial prevalence of the Delta variant remained below 5% of the total sequenced genomes, as it was outpaced by the Delta sub-lineages, AY.23 and AY.24. Two isolates exhibiting the Delta variant from lineage AY.23 were acquired from the isolation results. These isolates demonstrated vigorous development. The AY.23 mutation emerged in Indonesia and exhibits parallels to the Delta Plus variant identified in Britain.22 The AY.1 sub-lineage of the delta variant, distinguished by an additional K417N mutation in its spike protein, has been reported in England. The AY.1 variant, also known as the Delta Plus variant, is thought to exhibit the highest mortality rate. The Delta Plus form is thought to exhibit improved antibody evasion due to the K417N mutation. The Beta variant has been previously documented to possess a K417N mutation. Furthermore, relative to other Variant of Concerns (VoCs), the Delta plus variant has an increased propensity for transmission and a pronounced affinity for lung epithelial cells.23
Tables 2 and 3 present the nucleotide and amino acid mutations of the structural proteins of selected SARS-CoV-2 isolates. T19R increases the affinity to the ACE2 protein and facilitates its evasion of monoclonal antibodies, including regdanvimab and bamlanivimab, by reducing their binding to the virus.24 The Delta variant’s unique triple mutation, E156/F157/R158G, was precisely detected in our consensus nucleotide sequences. Nonetheless, the COVID-19 genome annotator program inadequately identified this non-codon-aligned triple mutation.25 The L452R mutation increases transmission, significantly intensifies severity, and facilitates the evasion of neutralizing antibodies generated by vaccines.26–28 The T478K mutation increases transmissibility and results in heightened disease severity. Furthermore, it diminishes the virus’s susceptibility to neutralizing antibodies generated by vaccines.29,30 The P681R mutation increases transmission, does not significantly impact disease severity, and partially diminishes vaccination efficacy by evading neutralizing antibodies.29,30 The P681R mutation increases transmission, does not significantly impact disease severity, and partially diminishes vaccination efficacy by evading neutralizing antibodies.31 The D614G mutation substantially enhances transmission rates, exerts no significant impact on disease severity, and markedly diminishes vaccine efficacy. The vaccine resulted in a 3-6-fold reduction in the titer of serum neutralizing antibodies (NABs) against the pseudovirus.32,33
The D950N mutation in the spike protein of Delta variant was identified in a region external of the receptor-binding domain (RBD), which is recognized for its stability and resistance to recurrent mutations.34 The V1264L variant in the spike protein introduces an acidic dileucine motif. This motif may affect the endocytosis process for several receptors associated with the resulting protein.35–37 V1264L possesses the capability to enhance the functionality of the spike protein. The presence of D63G was associated with a notable increase in viral load and may enable the virus to evade the immune system during replication.38
The mutation in the M gene confers a greater biological fitness benefit, potentially associated with glucose absorption during viral replication. Consequently, it is essential to integrate this lineage into current genomic surveillance efforts and meticulously evaluate its possible effects on heightened pathogenicity and treatment considerations.39 The mutations D63G, R203M, G215C, and D377Y in the N gene were associated with the highest incidence of intensive care unit admissions attributed to the Delta variation. Alterations were detected in an instance of breakthrough reinfection, in which the patient had hypoxia and necessitated hospitalization. These mutations were significantly associated with mortality, as detailed in a study.40 These modifications may facilitate the propagation of the virus.
Numerous Omicron lineages have been identified following the classification of B.1.1.529 as a Variant of Concern (VOC) on November 26, 2021. The Omicron variant has been categorized into several sub-lineages, one of which is BA.11. The Omicron sub-variants demonstrate an increased transmissibility relative to the Omicron variant (BA.1) and Delta. Nonetheless, an Omicron infection is less severe than a Delta infection. The BA.5 subvariant of Omicron is presently the most transmissible subvariant and has emerged as the predominant variant in circulation in the United States and numerous other regions globally. BA.5 is gradually replacing the original BA.1 and BA.2 Omicron subvariants of SARS-CoV-2 in several countries.40 The Omicron BQ.1 subvariant is swiftly becoming as the predominant variant in numerous countries globally, despite its recent emergence. Initial research indicates that BQ.1 is a subvariant of BA.5, with many progeny including BQ.1.1, BQ.1.2, BQ.1.3, and BQ.1.4.2. Alongside BQ.1, its progeny BQ.1.1 is also being monitored with increasing prevalence globally. The distribution of BQ1.23 is as follows: the United States of America comprises 26.0%, Australia 12.0%, Indonesia 9.0%, Japan 7.0%, and Germany 5.0%.41
We observed that the isolated SARS-CoV-2 omicron variant exhibited a mutation at T91 in the E protein. The T9I mutation results in the formation of a nonselective ion channel with reduced sensitivity to acid. T9I also decreased cytokine production and mitigated cell death. The alterations in channel properties may account for the Omicron variant’s diminished efficacy and lower extent of induced cellular damage. The severity of ionic dyshomeostasis, membrane disruption, and lysosome-related cell death would be mitigated by the enhanced chloride influx and reduced potassium efflux facilitated by T9I channels.42
Alterations Q19E and A63T were identified in all major Omicron subvariants. The closeness of the A63T mutation to a key location indicates that it may influence the stabilization of the M protein dimer.42 The N-terminal D3G and D3N mutations were solely identified in BA.1 and BA.5, respectively, possibly resulting in the establishment of the N-myristoylation site at positions 3–8.43 The P13L mutation may enable the virus to evade cellular immunity.44 R203K mutations augment the transmission and severity of particular SARS-CoV-2 variants.45 The Alpha and Gamma VOCs display modifications identical to the R203K and G204R mutations in the N protein, which may result in increased viral loads and augmented expression of subgenomic RNA.46,47
It was determined that several isolates are available for the development of next-generation vaccines targeting distinct SARS-CoV-2 variants, including Alpha, Delta, and Omicron. Sequencing test results for nine isolates indicated that four samples were identified as the Alpha (2) and Delta (2) variants, lineage B.1 and AY.23, respectively, while five samples were identified as the Omicron variant, with lineages BA.1.1, BA.5.2, and BQ.1.23.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)