Keywords
Ichthyosporea, small RNA, microRNA, miRNA, origin of animals, evolution, gene regulation
This article is included in the Genomics and Genetics gateway.
Ichthyosporea, small RNA, microRNA, miRNA, origin of animals, evolution, gene regulation
microRNAs (miRNAs) are short RNA molecules, 21-26 nucleotides, which post-transcriptionally regulate other genes.1,2 They are generated from genome-encoded single-stranded hairpins and are involved in a variety of physiological processes such as embryonic development, cell proliferation, immune response, and stress response.2
miRNAs have been identified across a range of eukaryote lineages, including plants, animals, green and brown algae, slime molds, and excavates.2–4 Among animals, miRNAs have been extensively studied in bilaterians, but also identified in cnidarians and sponges,5,6 two phyla whose ancestors evolved before the emergence of Bilateria. More recently, miRNAs have also been discovered in unicellular relatives of animals, the ichthyosporeans, but only in a single genus, Sphaeroforma.7
Despite these discoveries, the diversity and functional roles of miRNAs in ichthyosporeans, sponges, and cnidarians remain poorly understood. It is unclear how miRNAs emerged and evolved within Holozoa (animals and their closest unicellular relatives) and how their biogenesis and functions diversified across these lineages. And while bilaterian animals exhibit highly complex miRNA regulatory networks, little is known about how miRNAs contribute to gene regulation in earlier-branching holozoans.
In animals, miRNAs are generated from hairpin structures of RNA (pri-miRNAs) which are transcribed from the genome by RNA polymerase II.2 The pri-miRNA hairpin is cleaved inside the nucleus by the Microprocessor complex which consists of the proteins Drosha and Pasha (called DGCR8 in vertebrates). The resulting ~60 nucleotide precursor miRNA (pre-miRNA), which has an overhang of two nucleotides from the processing of the two RNase III domains in Drosha, is then exported to the cytoplasm. Here, the pre-miRNA structure is further cleaved by another double-RNase III protein called Dicer near the loop region of the pre-miRNA. This results in a double-stranded short RNA molecule with the characteristic 3′ two-nucleotide overhangs on both ends. One of the strands of this so-called miRNA duplex will be bound by the protein Argonaute (AGO). This strand is known as the mature miRNA (often denoted miRNA) and the other strand, which is released, is known as the star miRNA (often denoted miRNA*). These structural features, shaped by the sequential enzymatic actions, provide critical markers for identifying miRNAs in genomic and transcriptomic datasets.
The initial clues about the function of miRNAs in animals came from the observation of several conserved sites of lin-4 in C. elegans8 which were complementary to the 3′UTRs that had been proven to be essential for the repression of lin-14.9 We now know that miRNAs regulate gene expression by targeting specific gene transcripts in both plants and animals, however, they achieve it differently. While in plants, the miRNAs bind with a perfect, or near perfect complementarity to mRNAs (usually in the exons), animal miRNAs usually target mRNAs by a short complementary “seed” sequence (and most often target the 3′-UTRs).4,10 And in plants, as opposed to animals, this targeting mode generally results in the cleavage of the mRNA target.2 The cleavage of the mRNA usually occurs between nucleotide 10 and 11 of the miRNA and is performed by the PIWI domain in AGO.11
Until recently, the cleavage of mRNAs through AGO was believed to be limited to plants. However, a study on the cnidarian Nematostella vectensis found that many miRNAs actually target mRNAs via nearly perfect complementarity, and that these mRNAs are cleaved at position 10 in the target site, resembling the targeting mode in plants.12 It has been proposed that the “seed-based" targeting was a bilaterian innovation, which widened the binding capacity of miRNAs and the regulatory networks and contributed to the complexity of Bilateria.13 The finding of Moran et al.12 could therefore support cleavage as the ancestral function of animal miRNAs. However, the targeting modes of miRNAs in other basal holozoans remain entirely unknown, hence also whether cleavage-based targeting is a shared ancestral trait of all Holozoa or if it evolved independently in certain lineages.
The main objective of this study was to explore the presence, diversity, and functional roles of miRNAs in Ichthyosporea. Although miRNAs have previously been identified in the ichthyosporean genus Sphaeroforma, it remains unclear whether this genus is unique or if miRNAs are more widely distributed across other ichthyosporean lineages. To address this, we analyzed transcriptomic datasets from Abeoforma whisleri and Pirum gemmata, alongside new data from several additional species of Sphaeroforma. Additionally, we sought to determine whether miRNAs in S. arctica target and cleave mRNAs in a manner similar to the plant-like mechanism observed in the cnidarian Nematostella vectensis.
We studied six species of Ichthyosporea: Abeoforma whisleri (Awh), Pirum gemmata (Pge), Sphaeroforma arctica (Sar), S. sirkka (Ssi), S. tapetis (Sta), and S. gastrica (Sga). All species were cultured in tissue culture TC flasks T25 (Sarstedt, NRW, Germany) with 15 ml sterile Marine Broth (cat. no: 279110, Difco BD, NJ, US; 37.4 g/L) at 12°C with no light. Cultures were sustained monthly by transferring 1 ml of old cultures to 14 ml of Marine Broth.
Total RNA was isolated from the different species by first lysing the cells on a FastPrep system (MP Biomedicals, Santa Ana, CA, USA) followed by smallRNA purification using the mirPremiere RNA kit (cat. no: SNC50, Sigma-Aldrich, St. Louis, MO, USA). All cultures were saturated and contained a mix of different cell stages. For S. arctica we also included data from a time series experiment (in two biological replicates) where both small RNAs and mRNAs had been Illumina (Illumina, San Diego, CA, USA) sequenced at 12 time points starting from inoculation of an old culture and until 72 hours.14
The smallRNA libraries were prepared for sequencing with the NEBNext Small RNA Library Prep kit (cat. no: E7330L, New England Biolabs, Ipswich, MA, USA) and sequenced on the Illumina NextSeq 500 platform with 75 bp single end reads. Library preparation and sequencing services were provided by the Norwegian Sequencing Center (NSC).
The resulting Illumina reads were first trimmed for sequencing adaptors and low-quality bases using Trimmomatic v0.3815 (parameters: ILLUMINACLIP:TruSeq3-SE.fa:2:25:7 SLIDINGWINDOW:4:28 LEADING:28 TRAILING:28 MINLEN:18) by removing bases with a phred score less than 28 from both 5′- and 3′ ends. In addition, a sliding window of 4 nucleotides was used, trimming sequences from the 5′ end if the average phred score dropped below 28. Reads shorter than 18 bp were filtered out. Next, we used PrinSeq-lite v.0.20.416 to filter reads containing N′s and reads longer than 26bp (parameters: -max_len 26 -ns_max_n 0).
DNA was isolated from Sta, Sga, Awh and Pga using the DNeasy Blood & Tissue Kit (Qiagen, MD, USA) after lysing the cells using FastPrep as described above. All cultures were saturated and contained a mixture of cell stages.
For Sta and Sga, DNA was shipped to NSC for library preparation and sequencing. 150 bp paired-end (PE) libraries were generated and sequenced on one lane on the Illumina HiSeq4000. The resulting reads were processed into high-quality reads as described for the RNA data above. DNA from Awh and Pge was sequenced on the PacBio RS II instrument (Pacific Biosciences, CA, US). Libraries were prepared using the Pacific Biosciences 20 kb library preparation protocol, and size selection of the final libraries was performed using a BluePippin (Sage Science, MA, US) with a 7 kb cut-off. The libraries were sequenced on 6 SMRT cells. For Awh and Pge we also downloaded publicly available Illumina data from NCBI (BioProject PRJNA360047). The PacBio reads were used as they were delivered from the sequencing facility, taking advantage of the error correction through the use of Illumina reads in the hybrid assembly described below.
For all species, draft genome assemblies were created using Spades v.3.13.17 For Awh and Pge we used hybrid assembly with both Illumina and PacBio reads. As the assemblies were only used to identify precursor miRNAs, we did not perform scaffolding or other optimizations aimed at generation as long, or complete, genomic fragments as possible.
For Sar and Ssi we downloaded the already published genome assemblies (NCBI accessions GCA_008580545.1 and GCA_001586965.1).
We sequenced the expressed transcriptomes of Awh, Pge, Ssi, Sta and Sga by isolating total RNA using the RNeasy Mini Kit (Qiagen, MD, USA) after cell lysis using FastPrep as described above. All cultures were saturated and contained a mixture of cell stages. The purified total RNA was sent to the NSC for library preparation and sequencing. RNA from Awh, Pge, Sta and Sga was prepared into 150 bp PE libraries and sequenced together on one lane on the Illumina HiSeq X platform. RNA from Ssi was prepared into a 300 bp PE library and sequenced on an Illumina MiSeq.
The sequence reads were processed into high-quality reads using Trimmomatic as described above and assembled into contigs using Trinity v2.8.2.18 Protein coding sequences were predicted using TransDecoder v5.5.0.19
For Sar we downloaded the transcriptome assembly and annotation accompanying the genome assembly on NCBI (accession. GCA_008580545.1).
After trimming, the smallRNA reads were processed by the script process-reads-fasta.py which comes with the program miR-PREFeR.20 This script collapses identical reads into a single sequence with a number in the fasta header that indicates the number of identical reads. This was done to reduce the data size and speed up the analysis.
miRNAs were identified using a two-step process. First, miR-PREFeR was run to identify potential primary miRNA (pri-miRNA) sequences in the different genome assemblies. Then, the tentative pri-miRNAs were manually curated by inspecting the mapping of small RNA reads against the pri-miRNA sequences and comparing it against a set of criteria (described below). The curated sets of miRNAs were divided into three categories: high-, mid-, and low-confidence, based on the number of criteria fulfilled.
pri-miRNA identification by miR-PREFeR
To identify potential pri-miRNA loci in the genomes, miR-PREFeR was run with the processed small RNA sequences and a genome assembly as inputs. The program was run with the following parameters: PRECURSOR_LEN = 500, READS_DEPTH_CUTOFF = 2, MAX_GAP = 100, MIN_MATURE_LEN = 18, MAX_MATURE_LEN = 26, ALLOW_NO_STAR_EXPRESSION = N, ALLOW_3NT_OVERHANG = N. This means that the size of pri-miRNAs cannot be longer than 500nt, that at least 2 small RNA reads need to map to the mature region of the pri-miRNA and at least 1 read needs to map to the star region, that the length of the mature region should be between 18nt and 26nt, and that 3nt overhang between mature and star sequences is not allowed.
Curation of miRNA candidates
To further validate the miRNA identification by miR-PREFeR, and reduce the number of false positives, we mapped the processed smallRNAs to the pri-miRNA sequences identified by miR-PREFeR using Bowtie v1.1.221 with the parameters: -p 16 -v 0 -k 20 –best --strata --norc. This means that no mismatch is allowed, that up to 20 valid alignments are reported, that strand bias is eliminated and that no reads are mapped to the reverse complement. The read mapping was then visualized in Geneious v11.1.4 (www.geneious.com) and each miRNA candidate was inspected manually and scored according to the criteria determined by Kozomora and Griffith-Jones22 Fromm et al.,1 but slightly modified according to criteria updated by Axtell and Meyers23 (some of these criteria are also already fulfilled by the miR-PREFeR parameters) (see Table 1 and Figure 1).
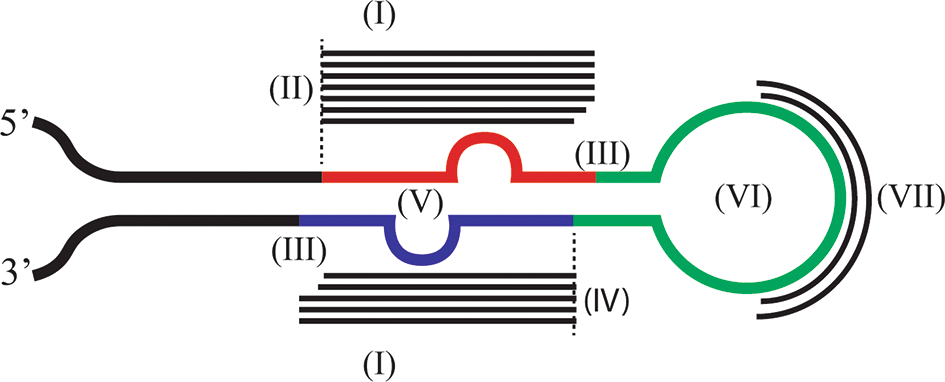
Legends: Schematic structure of a pri-miRNA hairpin as identified by miR-PREFeR and accepted as a valid pri-miRNA in this study. Red color indicates the mature region, blue color indicates the star region. The two bulges in the mature and star regions indicate non-complementary nucleotides. Green color indicates indicates the loop, and black color indicates the flanking regions. Thin black lines indicate smallRNA reads mapping to the pri-miRNA sequence. The dashed lines indicate the 5’ starting point of the mature and star regions. The roman letters in parentheses correspond to the different miRNA identification criteria in Table 1.
The miRNA candidates predicted by miR-PREFeR were divided into the following categories depending on how many of the above criteria they fulfilled (in addition to the settings already used by miR-PREFeR):
High-confidence miRNAs: miRNA candidates that fulfilled all seven criteria in Table 1. As an example, miRNAs in this category are named Sar-high-miR-1.
Mid-confidence miRNAs: miRNA candidates that fulfill six of the criteria. As an example, miRNAs in this category are named Sar-mid-miR-1.
Low-confidence miRNAs: miRNA candidates that fulfill five of the criteria. As an example, miRNAs in this category are named Sar-low-miR-1.
Discarded miRNAs: miRNA candidates that fulfill less than five of the criteria.
Some miRNAs were reported to arise from both strands of the same genomic locus, and with the same small RNA reads mapping to the mature and star regions. These cases could be due to artifacts in the genome assembly or from true inverted repeats. Nevertheless, we only counted these miRNAs as a single miRNA. And in cases where two pri-miRNA sequences were identical, but originated from different genomic loci, they were counted as two miRNAs.
Removal of non-coding RNAs
After curation, the miRNA precursor sequences were analyzed with the cmscan function of infernal v1.124 (parameters --cut_ga, --rfam, --nohmmonly, ---tblout) against the Rfam database25 to identify other types of non-coding RNAs that are not miRNAs. Precursors with significant matches were discarded from the final set of miRNA sequences.
Identification of evolutionary conserved miRNAs
We used the pri-miRNA sequences of the identified miRNAs in a pairwise blast against the genomes of the different ichthyosporeans. We used Blastn implemented in Geneious with the parameters: scoring (match mismatch) 2-3, maximum hits 10, max E-value 1e-10, Gap cost (open extend) 5 2, word size 7.
Genomic loci which covered the entire pre-miRNA (when a pri-miRNA had several matches in a genome, the match with the lowest E-value was selected) were selected to further investigate whether they were also transcribed and processed into a proper miRNA. To do this, the small RNA reads were mapped to the conserved genomic regions and evaluated using the criteria described above. Since these genomic loci are evolutionary conserved, and the query sequence already fulfill the criteria for a miRNA, we only required that these conserved genomic loci only fulfilled criteria I in Table 1 (i.e. that at least two reads map to the mature region and one read to the star) to designate it as a conserved miRNA.
miRNA target prediction
Tapir v1.226 was used to identify potential miRNA-targeted genes with near-perfect complementarity. Tapir was run using the mature miRNA sequences, as well as star sequences in cases where they were expressed equally high as the matures. In this case they were annotated as co-mature. The mature and co-mature sequences were searched against the assembled transcriptomes of the different ichthyosporean species. Tapir was run with the precise search option which selects potential targets according to a complementarity score cutoff (default 4) and an estimated minimum free energy (mfe) ratio of the miRNA:mRNA duplexes (default 0.7, which indicates that at least 70% of a miRNA:mRNA duplex should have a perfect complementarity to pass the threshold).
To annotate the potential target genes, protein sequences (for protein coding genes) were blasted against the RefSeq protein database using blastp with default settings, while the nucleotide sequences of non-coding genes were blasted against RefSeq RNA database with default settings.
To identify genes potentially targeted by miRNAs similar to that in animals, TargetScan v.7.027 was used with the “seed region” (nucleotides 2-8) of the mature and co-mature sequences as input and run against the 3′-UTRs of the Sar transcriptome genome with default parameters. There are four types of targeting sites identified by TargetScan 8mer-A1 (full match across the entire seed region and with an A nucleotide opposite of position 1 of the 3′ UTR), 7mer-m8 (match from position 2-8 of the seed), 7mer-A1 (match from position 2-7 in the seed and and A opposite position 1), and 6mer (match from position 2-7 in the seed), with decreased decreasing stringency and estimated effect on the targets.
Total RNA isolation
Degradome-seq was only performed on S. arctica. 1 ml of saturated Sar culture (containing mostly senescent cells) was transferred into 14 ml fresh Marine Broth. Total RNA was extracted 30h, 48h and 54h after inoculation (two biological replicates for each time points) using the RNeasy Mini Kit (Qiagen, Hilden, Germany), followed by DNase I (Invitrogen, CA, US) and RNA cleanup (Zymo research, CA, US) treatments according to provided protocols. The quality of the total RNA was evaluated on the Bioanalyzer 2100 (Agilent Technologies, CA, US).
Degradome library preparation and sequencing
The isolated total RNA was sent to LC sciences (Houston, TX, US) for preparation of the degradome libraries and Illumina sequencing. Briefly, the libraries were created by first capturing polyadenylated RNAs, then a sequencing adapter was ligated to the 5′-end of mRNAs. This adapter will only ligate to a 5′-monophosphate (i.e. uncapped 5′ ends). Then cDNA was generated using random primers and only those fragments carrying the 5′ sequencing adapter were sequenced. The cDNA fragments were sequenced from the 5′ sequencing adapter as 50 bp single-end reads on an llumina HiSeq 2500 instrument. The resulting reads were processed as described for the other small RNA reads above.
Identification of cleaved target genes
CleaveLand v4.528 was run with a p-value cutoff of 0.05 to report a significant miRNA-directed mRNA cleavage. CleaveLand first identifies potential binding sites for the mature miRNAs in the transcriptome, and then it aligns the degradome reads to the transcriptome and counts the number of reads starting at each nucleotide (degradome density). The peaks of reads starting at each position are divided into four categories, 0-4, where category 0 has the highest support for miRNA-directed cleavage. These peaks are then compared to the mature miRNA binding sites. If there is a peak exactly at the predicted cleavage site (between position 10 and 11 in the mature miRNA), a p-value is calculated, which takes both noise (the chances of observing the degradome peak of the given category at random) and the quality of the predicted target site (alignment score) into account. CleaveLand creates the degradome density by mapping with Bowtie (parameters: -f -v 1 --best -k 1 --norc -S). A program called GSTAr (distributed with CleaveLand) identifies the target sites by aligning the mature sequences. As input to CleaveLand both the mature (and co-mature) sequences of the miRNAs from all three categories of miRNAs from Sar and the Sar transcripts (including UTRs) were used.
The number of reads in the small RNA libraries from the different ichthyosporean species is described in Table 2. For miRNA identification we combined all the libraries from each species, hence the numbers in Table 2 represent combined libraries per species (for Sar this includes the time series datasets). Overall, removing low quality base calls and reads, as well as retaining only reads between 18-26 nucleotides removed on average close to 60% of the sequences (the bulk of the reads removed was due to the length filtering).
For Awh, Pge and Sar there was a clear enrichment of reads at 20 nt ( Figure 2). For the other species, the read lengths 18-26 nt was more evenly distributed, although 20 nt was the most common length except in Sga.
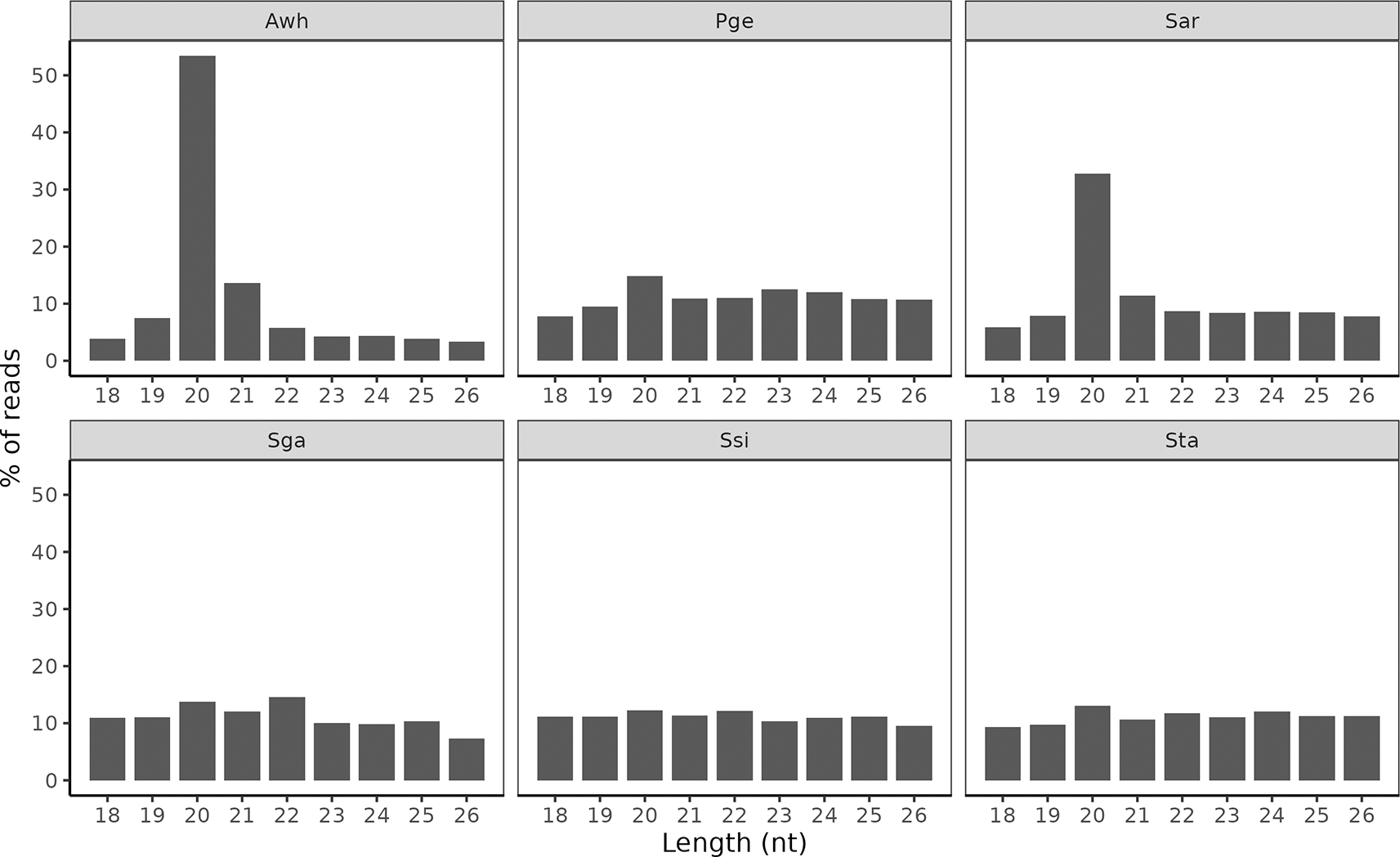
The fraction of reads with nucleotide lengths between 18-26 nt after processing.
Manual curation of the initial predictions made by miR-PREFEeR identified a total of 180, 74, 105, 13, 16 and 19 miRNAs in Awh, Pge,Sar, Ssi, Sta and Sga respectively ( Table 3). These miRNAs were further classified into three categories dependent on how confident we were in the predictions after mapping the small RNA reads to the precursor sequences (see Methods section and Figure 3 for examples of miRNAs from the three categories).

Legends: Examples of high-confidence (A), mid-confidence (B), and low-confidence (C) miRNAs from the different ichthyosporean species. The y-axis shows mapped reads per million processed reads (RPM), and the nucleotide position on the pri-miRNA sequence is shown on the x-axis. The secondary structures of the pri-miRNAs as predicted by mfold37 are shown next to the mapping. Reads mapping to the mature region are colored red and reads mapping to star region are colored blue. The same colors are used on the secondary structure.
Awh and Sar showed a clear A/U preference at the first nucleotide of miRNA mature strands ( Table 3). For the other Sphaeroforma species we detected very few miRNAs, so we cannot determine whether there was an A/U preference or not. For Pge no such A/U signal was detected.
Most of the identified miRNAs in all the ichthyosporean species were predicted to have at least one target according to Tapir ( Table 4). However, when considering only scores of 2.5 or lower (where a Tapir score of 2.5 may indicate complementarity along the entire miRNA with one seed mismatch, and a score of 0 represents a perfect complementarity), approximately 50% or fewer of the miRNAs had a potential target. It should be noted that these results are highly dependent on the number and quality of the transcriptomes used as potential targets. For Pge, Sga, Ssi and Sga the transcriptome assemblies were probably highly fragmented and redundant, reflected in a high number of transcripts for these species and therefore a low fraction of potentially targeted transcripts. Among Sphaeroforma, Sar had a lot more predicted targets even though fewer mRNA transcripts were used as input. However, this is most likely because of the large difference in the number of predicted miRNAs from these species.
The TargetScan analysis was only run on Sar since full genome annotation of the 3′UTRs was only available for this species. This analysis identified more than 100,000 potential seed binding sites within the 3’-UTRs ( Table 5). Some miRNAs were predicted to target many different locations of the same gene. This was especially the case for Sar-high-miR-15 which had 937 potential target sites in a total of 300 different genes. Among those genes, Sarc4_g19159T, Sarc4_g11565T, Sarc_g22056T, and Sarc4_23261T contained binding sites for Sar-high-miR-15 at more than 20 different locations. Therefore, this analysis should only be used as an indication that there are potentially a huge number of binding sites in the 3’ UTR regions of the different transcripts, but experimental evidence is needed to confirm actual binding and transcriptional regulation.
Conservation across species and over long time scales can be an indication of functionally important miRNAs, and in animals conserved miRNAs are generally functionally important.2 Therefore, we investigated whether any of the identified miRNAs were conserved between the investigated ichthyosporeans. There were several pri-miRNA sequences that had homologous loci in the genomes of two or more ichthyosporeans ( Table 6). Most of these however were only processed into proper miRNAs in one of the species.
However, 13 different miRNAs had conserved pri-miRNA sequences that were also expressed and processed into proper miRNAs in two or more species ( Table 7). Based on sequence similarity between the pri-miRNAs, these 13 miRNAs seem to belong to four different evolutionary conserved loci ( Table 7 and Figure 4).
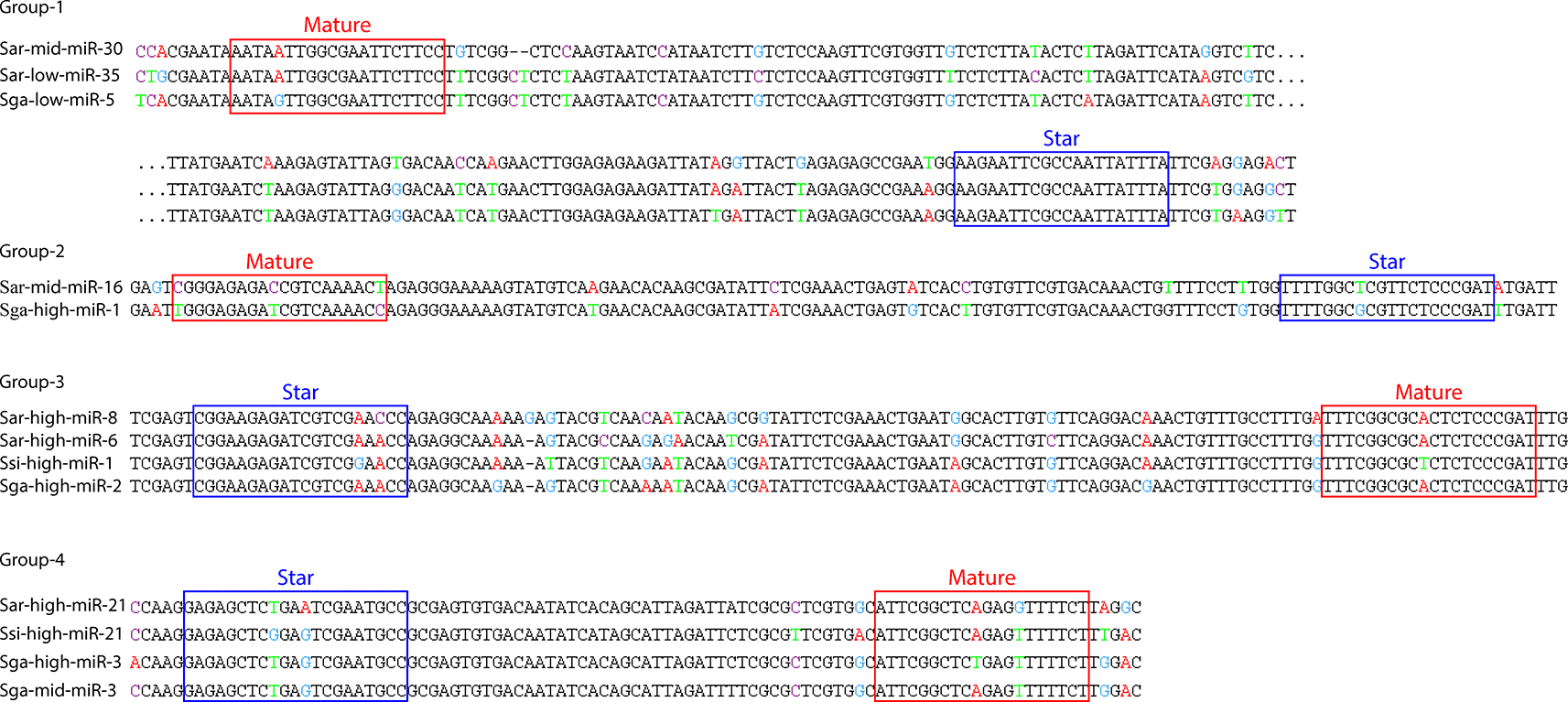
Legends: Pairwise MUSCLE alignment of the pri-miRNA sequences of the conserved pri-miRNA loci that were expressed and processed into proper miRNAs in two or more species. The red and blue rectangles indicates the mature and star regions. Colored nucleotides in the alignment indicate sites with sequence variation. The sequences in group 1 are wrapped and the dots indicate the break point. Group numbers correspond to the groups in Table 7.
In plants, mRNA transcripts targeted by miRNAs are silenced by cleavage between position 10 and 11 of the mature miRNA binding site.12,29 These mRNAs will therefore lack the 5′-cap and can be identified by a so-called degradome sequencing (degradome-seq). Degradome-seq targets mRNAs which lacks the 5′-cap and is a modified version of 5′-Rapid Amplification of cDNA Ends (RACE) and similar to a 5′-uncapped polyadenylated mRNA sequencing (also referred to as parallel analysis of RNA ends (PARE).
We performed duplicate isolations of total RNA from three different timepoints and prepared degradome-seq libraries. From the sequencing of cleaved mRNAs we received between 20-30 million reads after quality processing ( Table 8). Overall, the number of degradome reads was between 25 ~ 35 million before processing, while after the number of reads decreased to between 20 ~ 30 million.
| Library | Raw reads | Processed reads |
|---|---|---|
| 30h_1 | 36 343 891 | 30 046 849 |
| 30h_2 | 34 362 834 | 29 194 149 |
| 48h_1 | 28 891 639 | 24 013 478 |
| 48h_2 | 36 088 177 | 31 236 800 |
| 54h_1 | 23 173 814 | 19 547 675 |
| 54h_2 | 32 508 076 | 28 123 224 |
CleaveLand identified 18 different mRNA transcripts that were potentially cleaved through miRNA-directed cleavage by 11 different miRNAs ( Table 9). 10 transcripts were cleaved only at a specific time point, 4 were cleaved at two time points, while 4 were cleaved at all three time points. Only two of the miRNAs did not start with an A or U. 15 out of 18 of the cleaved targets were targeted in exons, while one cleaved by Sar-mid-miR-32 co-mature strand was targeted at 5′UTR, and the rest of the targets were non-coding RNAs. Only six of the 18 transcripts were also identified as targets by Tapir. Two of the highly conserved miRNAs (Sar-high-miR-6 (Sar-high-miR-8 has identical mature sequences so these are functionally equivalent) and Sar-mid-miR-16), and two miRNAs with conserved pri-miRNAs only (Sar-mid-miR-3 and Sar-high-miR-2) had induced target cleavage. Furthermore, all the cleaved targets with category 0 were targeted by the same miRNA (Sar-mid-miR-3) which had conserved pri-miRNA sequence ( Figure 5).
| miRNA | Conserved | 1st nt | Category | Target | Tapir | mRNA feature | Timepoint | RefSeq hit |
|---|---|---|---|---|---|---|---|---|
| Sar-mid-miR-3 | pri-miRNA | A | 0 | Sarc4_g15395T | Yes | Exon | 30h, 48h | No hit |
| Sar-mid-miR-32 (co-mature) | No | A | 3 | Sarc4_g2876T | Yes | 5'UTR | 30h, 48h | Transmembrane protein 35A-like (6e-06) |
| Sar-high-miR-2 | pri-miRNA | A | 3 | Sarc4_g26280T | No | Exon | 30h, 54h | No hit |
| Sar-high-miR-6(8) a | miRNA | U | 1 | Sarc4_g31131T | No | Exon | 48h, 54h | Autotransporter domain-containing protein (3e-04) |
| Sar-mid-miR-3 | pri-miRNA | A | 0 | Sarc4_g19705 | Yes | Exon | 30h | No hit |
| Sar-low-miR-38 | No | G | 3 | Sarc4_g24093T | No | Exon | 30h | H/ACA ribonucleoprotein complex subunit 1 (5e-47) |
| Sar-low-miR-41 | No | U | 3 | Sarc4_g3869T | No | Exon | 30h | Translocation protein SEC62-like (7e-28) |
| Sar-mid-miR-3 | pri-miRNA | A | 0 | Sarc4_g5854T | Yes | Exon | 30h | No hit |
| Sar-mid-miR-3 | pri-miRNA | A | 0 | Sarc4_g19698T | Yes | Exon | 48h | No hit |
| Sar-mid-miR-6 | No | G | 3 | Sarc4_g6144T | No | Exon | 48h | Isopenicillin N synthase family oxygenase (6e-55) |
| Sar-low-miR-16 | No | U | 2 | Sarc4_g8134T | No | Exon | 48h | Zinc finger X-linked protein ZXDB-like (3e-54) |
| Sar-high-miR-16 | No | A | 2 | Sarc4_g31551T | No | Exon | 54h | No hit |
| Sar-mid-miR-3 | pri-miRNA | A | 0 | Sarc4_g32950T | Yes | Exon | 54h | Aldehyde reductase (6e-65) |
| Sar-low-miR-27 (co-mature) | No | U | 4 | Sarc4_g7215T | No | Exon | 54h | Sortase B protein-sorting domain-containing protein (3e-09) |
| Sar-mid-miR-16 | miRNA | U | 3 | Sarc4_g182T | No | Exon | 30h, 48h, 54h | Conserved hypothetical protein (6e-08) |
| Sar-mid-miR-16 | miRNA | U | 3 | Sarc4_g31933T | No | Exon | 30h, 48h, 54h | Putative E3 ubiquitin-protein ligase, makorin-related (6e-16) |
| Sar-mid-miR-3 | pri-miRNA | A | 0 | asmbl_10212 | No | Non-coding RNA | 30h, 48h, 54h | No hit |
| Sar-high-miR-2 | pri-miRNA | A | 3 | asmbl_14039 | No | Non-coding RNA | 30h, 48h, 54h | No hit |
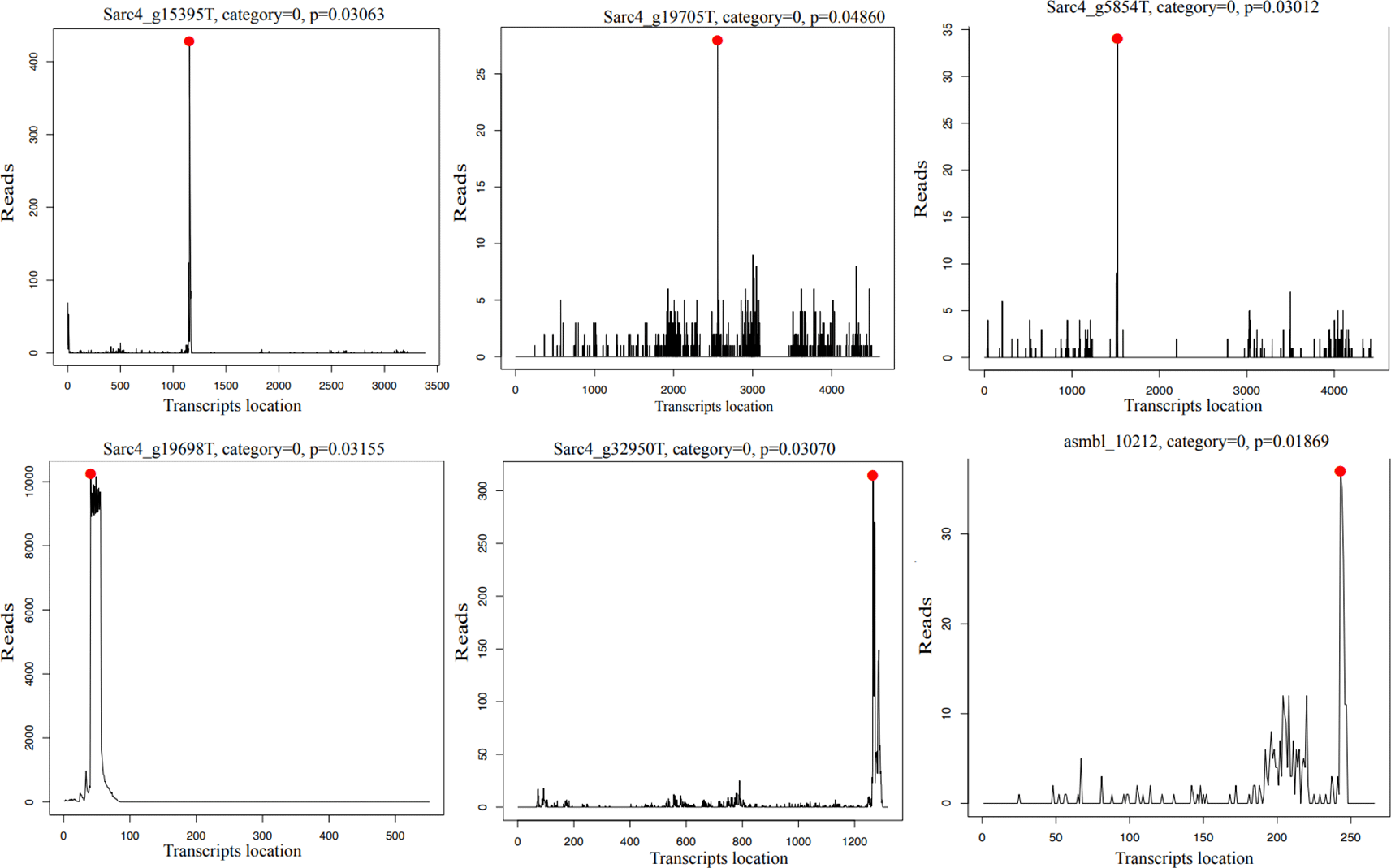
Legends: The plots show the category 0 targets identified by CleaveLand. The y-axis shows the number of degradome reads (from cleaved mRNAs) starting at each nucleotide position on the transcripts (x-axis). The red dots mark the degradome reads starting at the predicted cleavage site (i.e., at the 10th nucleotide of the complementary sequence to the mature miRNA).
We have identified miRNAs in two ichthyosporean species outside Sphaeroforma, A. whisleri and P. gemmata, as well as several novel miRNAs among Sphaeroforma sp. This extends the results of Bråte et al.,7 and shows that Sphaeroforma is not an exception, but that miRNAs are probably a general phenomenon among Ichthyosporea. This also strengthens the hypothesis that miRNAs were present in the last common ancestor of Holozoa, and that they have been lost in the lineages Filasterea, Choanoflagellata and Ctenophora.
Most of the identified ichthyosporean miRNAs are lineage-specific, meaning that they are not conserved between A. whisleri, P. gemmata and Sphaeroforma sp. This lack of conservation is perhaps unsurprising, given that these lineages are estimated to have diverged more than 500 million years ago.30 This suggest that most of the miRNAs evolved independently in each genus, and that there is a rapid birth-and-death dynamic in the evolution of miRNAs in Ichthyosporea. Similar patterns have also been observed in sponges and cnidarians, where no miRNAs are conserved between the major sponge lineages,6,31 and only a handful of the nearly 100 miRNAs in Nematostella vectensis are shared with Hydra, another cnidarian.12
Despite the lack of conserved miRNAs across Ichthyosporea, several miRNAs are conserved between the Sphaeroforma species. These species have very similar cellular morphologies and life cycles, and exhibit high similarity based on 18S small ribosomal subunit comparisons,32 although precise estimates of their divergence times are unavailable. Given their close relationships, an even higher degree of miRNA conservation across Sphaeroforma species might have been expected. The low observed conservation is likely due to incomplete identification of miRNAs in S. sirkka, S. tapetis, and S. gastrica because of poor quality of the transcriptome assemblies.
Additionally, there are many more primary miRNA genomic loci conserved between Sphaeroforma species than there are miRNAs. Even a few shared between S. arctica, A. whisleri, and P. gemmata. This suggests that lineage-specific miRNAs may have evolved from ancestral genomic loci that gained the ability to be transcribed, likely through the acquisition of transcription factor binding sites. This aligns with the idea of rapid evolutionary turnover in miRNA processing,33 where minor genomic changes can enable or disrupt miRNA expression and processing.
Because miRNAs are short non-coding molecules, often with very few nucleotides of functional importance, they can be difficult to identify based on sequencing and gene prediction alone. It is also obvious that miRNA identification is dependent on the input data. For example, the reason why so many novel miRNAs in Sphaeroforma are detected here compared to the 8 miRNAs identified in Bråte et al7 is probably because a much higher number of small RNA reads were sequenced, including different cellular stages. And in S. sirkka, which is very closely related to S. arctica with almost identical cellular morphology,32 only a few miRNAs were identified. But S. sirkka also had the least number of small RNA reads. However, the number of conserved pri-miRNAs between the two species is quite high and probably more of these could be identified as expressed had we sequenced the small RNA fraction deeper. For S. tapetis and S. gastrica we also identified few miRNAs despite a large number of small RNA reads. But these species have very poor and highly fragmented genome assemblies, generated from low coverage short read data. And this has probably reduced the power to detect miRNA precursor sequences in the genomes.
To identify novel miRNAs, one of the biggest jobs is to separate true miRNAs from other RNA segments with hairpin characteristics or other miRNA-resembling features.1 All miRNA identification tools report a large number of false positives, and even the accuracy of the best tool rarely surpasses 60%.23 Because Ichthyosporea are non-model species, with very little information available on the kind of characteristics to expect from the miRNAs, we used a less stringent system for miRNA identification but classified the miRNAs into three different confidence categories based on the number of identification criteria they met. This classification of miRNAs is by no means perfect, and there are several caveats; Of the seven identification criteria used here, we consider criteria I-IV the most important, as they directly reflect signatures of the pri- and pre-miRNA processing. Therefore, some of the miRNAs included here could be for instance either endogenous siRNAs or other spuriously transcribed and processed hairpin structures. This is especially true for the low-confidence miRNAs, where several of the miRNAs have uneven processing of the mature and star regions, or many reads mapping to the loop region. On the other hand, several low-confidence miRNAs do show consistent processing of the mature and star region, but could have unexpected hairpin structure, or some would lack star consistency but otherwise shown consistent mature processing. Therefore, it is not easy to simply discard the miRNAs belonging to the low- or even mid-confidence categories. Nevertheless, there are many miRNAs identified here belonging to the high-confidence category, and that these show all the signs of processing by the Microprocessor and are likely to be functional miRNAs.
The functional mechanisms of miRNAs in Ichthyosporea are unknown. It is unclear whether they operate like plant and cnidarian miRNAs, which target mRNAs through long complementary binding sites to cleave transcripts, or like bilaterian miRNAs, which primarily use a short seed region for target recognition. We investigated the possibility of animal-like targeting in S. arctica using TargetScan,27 which identifies potential targets based on seed region complementarity. This showed that thousands of transcripts contain binding sites for the miRNAs in their 3’ UTRs, indicating a potential for extensive gene regulation similar to bilaterian animals. However, the reliability of this prediction alone is very limited due to the small sequence length of seed regions and the lack of experimental validation. These results should therefore only serve as a foundation for future experimental studies.
Moran et al.12 showed that in Cnidarians miRNAs can induce mRNA target cleavage and speculated that this might be the ancestral mode of action for animal miRNAs. We examined whether this could also be the case for ichthyosporean miRNAs. Using Tapir,26 we identified thousands of mRNA transcripts across Ichthyosporea with high sequence complementarity to miRNAs, suggesting that many mRNAs could be targeted in this manner. This is further supported by our comparison of mRNA cleavage positions with the predicted target sites of the S. arctica miRNAs. We find clear indications for miRNA-induced slicing of mRNA transcripts for 11 of the predicted miRNAs. Many of these cleaved transcripts were targeted by miRNAs conserved between Sphareoforma sp. In bilaterians, evolutionarily older and conserved miRNAs tend to target conserved genes.2 In S. arctica, no such clear pattern is observed. Many of the cleaved transcripts targeted by conserved miRNAs are a mixture of genes with identifiable homologs in other species and apparently species-specific genes.
A notable feature of the miRNAs directing cleavage in S. arctica is their preference for starting with an A or U nucleotide. This is similar to what has been found for vertebrate miRNAs, where almost 90% of the miRNAs start with a U or an A nucleotide,1 and is believed to reflect the binding preference of the Argonaute protein34 in the formation of the gene silencing complex. Ichthyosporeans also have homologs of Argonaute,7 and the A/U bias may suggest that these miRNAs are also bound by Argonaute, although this remains to be verified.
It is worth noting that the degradome sequencing approach employed here likely underestimates the true number of cleaved targets. This is because mRNAs marked for cleavage often lose their poly(A) tail, and the poly(A)-selection step in the degradome-seq protocol would exclude such transcripts. Moran et al.12 demonstrated that using 5′-RACE, which sequences 5′-start sites independent of poly(A) selection, can identify significantly more cleavage events. Additionally, the CleaveLand28 algorithm used here only detects cleavage between positions 10 and 11 of the miRNA binding site. However, miRNA-induced cleavage in S. arctica could also occur at alternative positions. These factors suggest the number of cleaved targets in S. arctica is likely higher than identified in this study.
Thus, it is likely that ichthyosporean miRNAs bind to and regulate a diverse array of gene transcripts, though the precise targeting mode and functional outcomes remain to be experimentally verified. These findings indicate that S. arctica miRNAs regulate gene expression by inducing mRNA cleavage, a mechanism reminiscent of cnidarians and supporting the hypothesis that cleavage was the ancestral function of animal miRNAs. It remains to be determined whether such cleavage events occur at specific developmental time points, potentially linking miRNA activity to particular cellular stages.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)