Keywords
ShinyApp, Single cell, scRNA-seq
This article is included in the Bioinformatics gateway.
ShinyApp, Single cell, scRNA-seq
Single-cell RNA sequencing (scRNA-seq) technologies have revolutionized molecular biology by enabling transcriptome profiling at single-cell resolution.1–4 Additionally, substantial efforts have been made to develop computational pipelines and tools to improve the analysis and visualization of large scRNA-seq data sets. These advances have driven significant innovation in computational biology of over 1500 tools and various frameworks and software repositories, such as Bioconductor, Seurat, and Scanpy.5–9
As the community continues to publish and deposit single cell datasets, the need to facilitate their sharing among collaborators has become increasingly important. Web-based and open-source frameworks have emerged as valuable tools for sharing results of single cell experiments with both intramural and extramural collaborators. For instance, stand-alone software such as iS-CellR, ASAP, SingleCAnalyzer, ShIVA, singleCellTK provide graphical interfaces for single cell RNA-seq datasets.10–14 Other open-source methods based on the R shiny framework such as ShinyCell, SCHNAPPs, SeuratV3Wizard, and iSEE have become a free-to-use solution to share results for computational biologists.15–18 With the increasing adoption of single-cell experimental approaches generating more complex data, it is necessary to improve methods of data sharing to accommodate the growing complexity of single-cell datasets.
Taking advantage of the Shiny framework (https://shiny.rstudio.com/), here we present ShinyCellPlus, an improved Shiny/R application based on ShinyCell16 and R/Bioconductor packages as ggvolc, ggplot2, and scToppR,19 to provide interactive visualizations of single cell RNA-seq results with specific and upgraded functionalities. ShinyCellPlus is an exploratory tool that allows scientists to share single cell data with collaborators or the scientific community. The R package and code are available at https://github.com/BioinformaticsMUSC/ShinyCellPlus.
ShinyCellPlus was developed to expand the capabilities of the ShinyCell package, enabling more streamlined visualization of single-cell RNA-seq data. It achieves this by deploying a Shiny web app that can be used either on specific websites or locally. ShinyCellPlus relies on a Seurat object containing relevant single-cell genomic data and metadata files with all the necessary variables for visualizations. To maximize the app's functionality, we recommend converting SingleCellExperiment objects into Seurat objects before deployment. The user interface (UI) is organized with tabs and modules at the top, while specifications, thresholds, and other visualization options are located on the left-hand sidebar, preserving the original ShinyCell UI structure. To enhance the ShinyCell package, we incorporated additional miscellaneous data into the Seurat object based on single-cell analysis. This allows users to quickly identify cell-type markers, examine genes differentially expressed by cell type, and generate intuitive visualizations (Fig. 1) that can be downloaded in different formats. The Shiny app and visualization methods are compatible with modern browsers and have undergone testing on Google Chrome (v98.0.4758.80) and Firefox (v96.0).
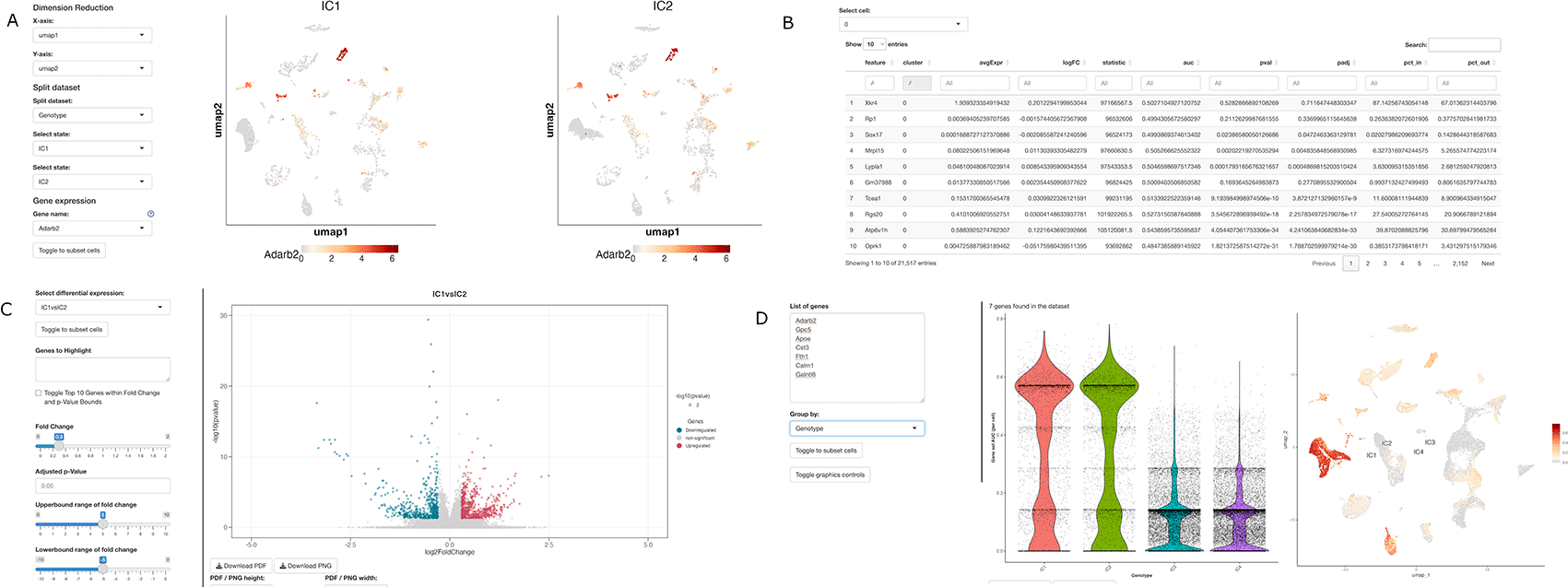
(A) Example of the “split-by” new sections.
(B) Visualization of the markers table new sections. Similarly, two additional sections report the differential expression tables based on the analysis performed.
(C) Visualization section for the genes differentially expressed. Volcano plot can be modified using the left section with visualizations and thresholds parameters.
(D) Visualization section for the gene enrichment based on AUCell analysis.
To facilitate comparative visualizations, ShinyCellPlus includes specific functionalities to split the Seurat data by sample or conditions.
• Gene Dual Coexpression - Added a second interactive dimensional reduction plot to express the relationship between two genes along with the first plot.
• Split Dataset: Cell Info - Allows for comparison of conditions and other categorial data stored in the Seurat metadata.
• Split Dataset: Gene Expression - Allows for comparison of conditions stored in the Seurat metadata with selection of gene expression on both graphs.
• Split Dataset: Gene Coexpression - Allows for comparison of conditions stored in the Seurat metadata with selection of two genes for coexpression on both graphs.
ShinyCellPlus enables the interactive visualization of marker genes and differential expression (DEG) statistics. To achieve this, ShinyCellPlus requires a tab-separated input for both markers and DEGs. To calculate markers and differential expression, we provided two examples based on the R packages Presto (https://github.com/immunogenomics/presto), Libra (https://github.com/neurorestore/Libra), or FindMarkers() function in Seurat. Presto uses an optimized Wilcoxon test to compute cell-type or cluster markers, and the resulting tables can be stored in the designated section called “misc” within a new Seurat object. Differential expression (DEG) statistics can be computed using the run_de() function from the R package Libra or the FindMarkers() function from the R package Seurat. To streamline the incorporation of the resulting tables into the Seurat object, we have developed a function called add_libra_DE_table_to_seurat(), which can be found in our GitHub repository (https://github.com/BioinformaticsMUSC/ShinyCellPlus). The Marker table and DEG table are presented interactively, with a drop-down menu that allows users to select cell types and different DEG tables from various comparisons. For the display of interactive tables, we utilize the R package DT (https://rstudio.github.io/DT/). Additionally, we offer an interactive volcano plot for a visual exploration of DEG statistics as well as interactive balloon and cluster dot plots visualizing gene ontology queries from ToppGene (https://toppgene.cchmc.org/). The volcano plot is created using the R package ggvolc (https://github.com/loukesio/ggvolc), and the gene ontology balloon and cluster dot plots (Fig. 2) are made by using R package scToppR (https://github.com/BioinformaticsMUSC/scToppR/), an API for ToppGene.

(A) Cluster ontology dotplots; to allow for a large number of clusters, the canvas’ height and width are adjustable, along with image exports height and width.
(B) Ontology balloon plot showing most significant ontological category for cluster.
For gene set signatures, ShinyCellPlus employs the R package AUCell (https://github.com/aertslab/AUCell).20 AUCell enables the identification of cells displaying active gene sets, such as signatures or gene modules, within single-cell RNA-seq data. AUCell statistics can be stored in a designated section called "aucell" within a new Seurat object. In ShinyCellPlus, users can input lists of genes to be plotted interactively, allowing them to quickly visualize relevant gene modules.
An RStudio environment is recommended for ease of running the Shiny output files locally. The package’s DESCRIPTION file contains ‘Depends’ and ‘Imports’ lists for all external packages used within the creation of the Shiny app files as well as what is required when running said files through Shiny. Runtime can be memory intensive depending upon the data contained within the initial input Seurat object. Often there is lag from when the app’s HTML initially loads to when the first graphs appear, depending upon the size of the associated data files, but subsequent graph updates are much quicker and responsive. Instructions for general operation of the package can be found within the GitHub’s README.
ShinyCellPlus stands out as an intuitive and graphical tool designed for single-cell RNA-seq, enhancing the already excellent ShinyCell R package. This advanced, user-friendly app, built on the widely adopted Seurat pipeline, provides a more sophisticated platform for single-cell analyses. Our team is committed to improving and expanding the capabilities of ShinyCellPlus in response of the growing interest in single-cell omics and friendly user's apps. It not only generates publication-ready plots and tables but also allows users to interactively explore single-cell data and intuitively examine individual genes of interest.
Source code available from: https://github.com/BioinformaticsMUSC/ShinyCellPlus
License: GPL-3.0 License
Archived source code at time of publication: https://doi.org/10.5281/zenodo.14713623
License: GNU General Public License v3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)