Keywords
MEN 1, hyperparathyroidism, neuroendocrine tumors, genetic counseling, Tc99 MIBI scans, multidisciplinary management.
This article is included in the Rare diseases collection.
MEN 1, hyperparathyroidism, neuroendocrine tumors, genetic counseling, Tc99 MIBI scans, multidisciplinary management.
See the authors' detailed response to the review by Pinaki Dutta
Multiple Endocrine Neoplasia Type 1 (MEN1), also known as Wermer’s syndrome, is a rare hereditary disorder characterized by the predisposition to develop tumors in multiple endocrine organs. MEN1 is caused by mutations in the MEN1 gene located on chromosome 11, which encodes the tumor suppressor protein menin. This syndrome primarily involves the parathyroid glands, endocrine pancreas, and anterior pituitary, although other tissues and organs may also be affected. MEN1 follows an autosomal dominant pattern of inheritance, with a prevalence of approximately 2 in 100,000 individuals. The condition poses significant diagnostic and management challenges due to its multisystemic involvement and variable clinical presentation.1,2 Parathyroid tumors are the most common manifestation of MEN1, occurring in over 90% of cases. These tumors often lead to primary hyperparathyroidism, which presents early, usually in the second or third decade of life, compared to sporadic hyperparathyroidism seen later in life. In MEN1-associated hyperparathyroidism, multiple glands are typically involved, and there is a higher rate of recurrence after surgery compared to sporadic cases. This condition often results in hypercalcemia, leading to symptoms such as bone pain, fractures, nephrolithiasis, and fatigue, further complicating the patient's clinical course. Gastroenteropancreatic neuroendocrine tumors (NETs) are another hallmark of MEN1, affecting up to 70% of individuals with the syndrome. These tumors, including gastrinomas, insulinomas, and non-functioning pancreatic NETs, often exhibit an aggressive clinical course and can result in severe symptoms such as refractory peptic ulcers, diarrhea, hypoglycemia, and abdominal pain. Despite advancements in imaging and biochemical tests, these tumors are often diagnosed late, when they have already metastasized, making management more complex.
Pituitary adenomas, which occur in approximately 15-90% of MEN1 cases, add another layer of complexity to the syndrome. While many are non-functioning, some secrete prolactin or growth hormone, leading to clinical manifestations such as galactorrhea, menstrual disturbances, acromegaly, or gigantism. These adenomas are often resistant to conventional treatment and may require surgical or medical intervention.3
The variable penetrance and expression of MEN1 make its diagnosis particularly challenging. Patients may initially present with symptoms from one organ system, such as recurrent kidney stones or refractory peptic ulcer disease, leading to delayed recognition of the underlying syndrome. Early diagnosis is critical, as MEN1-related tumors often follow a more aggressive course compared to their sporadic counterparts. Genetic testing for MEN1 mutations and screening of first-degree relatives are essential components of the diagnostic approach.
Management of MEN1 requires a multidisciplinary approach, involving endocrinologists, surgeons, radiologists, and oncologists. Treatment strategies are individualized, focusing on controlling hormonal hypersecretion, addressing tumor burden, and mitigating complications. Advances in imaging modalities, such as 68Ga-DOTATATE PET/CT, and novel therapeutic agents, including somatostatin analogs and molecularly targeted therapies, have improved the diagnostic and therapeutic landscape for MEN1 patients. However, the syndrome remains a lifelong challenge due to its high recurrence rates and the need for continuous surveillance.4
This report presents two cases of MEN1, highlighting the complexities of its diagnosis and management. Through these cases, we aim to provide insights into the unique challenges posed by this rare condition and emphasize the importance of early recognition and a coordinated, multidisciplinary approach to care.
Multiple Endocrine Neoplasia Type 1 (MEN1), also known as Wermer’s syndrome, is a rare hereditary condition that involves the development of tumors in endocrine glands such as the parathyroid, anterior pituitary, and pancreatic islet cells. This report discusses two distinct cases of MEN1, detailing their clinical presentations, investigations, and management.
Clinical Presentation: A 47-year-old unmarried female with a history of chronic kidney disease (CKD) and hypothyroidism presented with a five-day history of watery diarrhea, nausea, and vomiting. The diarrhea was waxing and waning in nature, and there was no associated fever, pain abdomen, or weight loss. She had a prior history of hospital admissions for similar complaints and was managed with IV fluids, antibiotics, and probiotics, which showed minimal improvement.
On examination, she was noted to have mild pallor and clinical signs of dehydration. No lymphadenopathy or hepatosplenomegaly was observed. The physical examination of the abdomen was unremarkable.
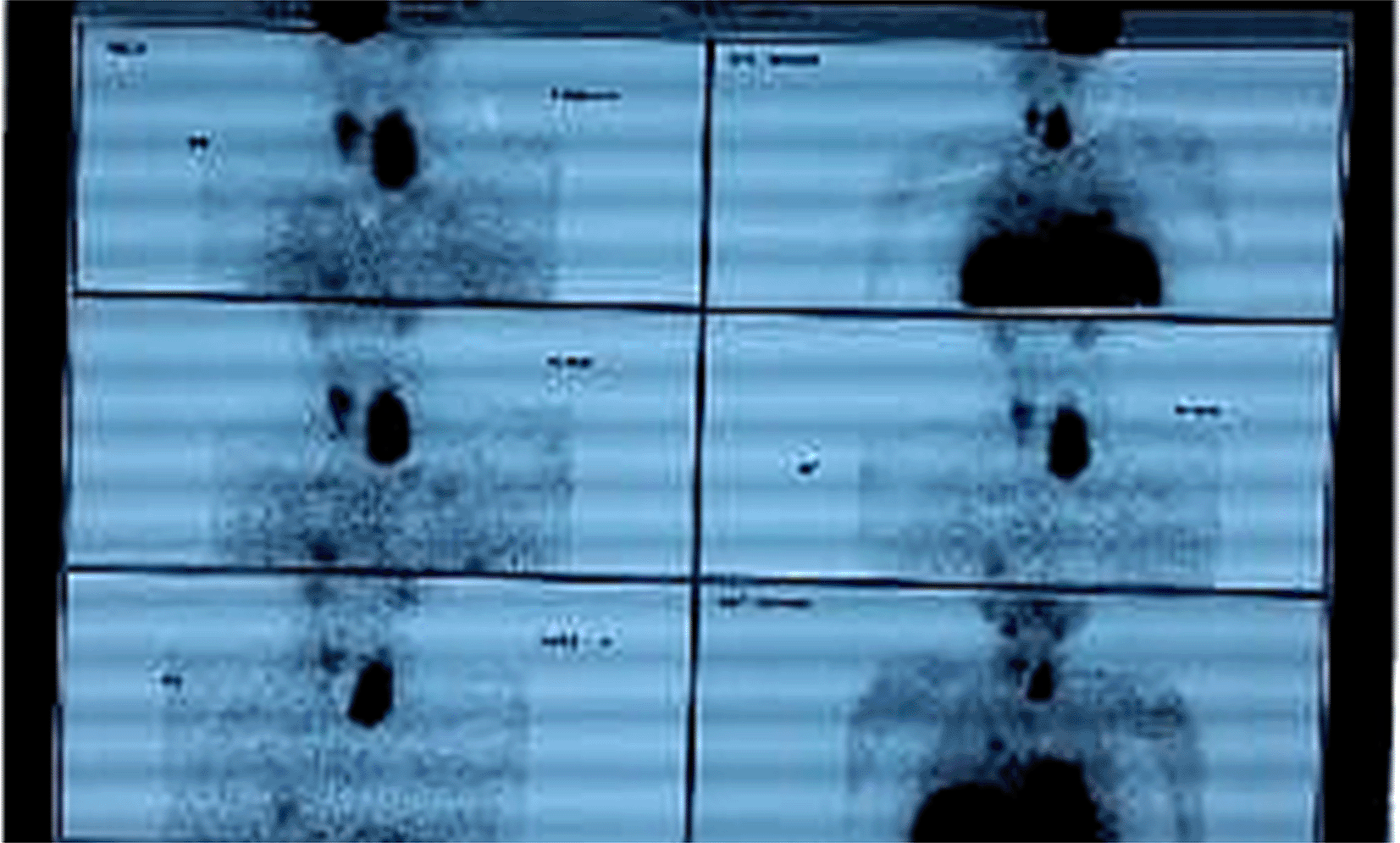
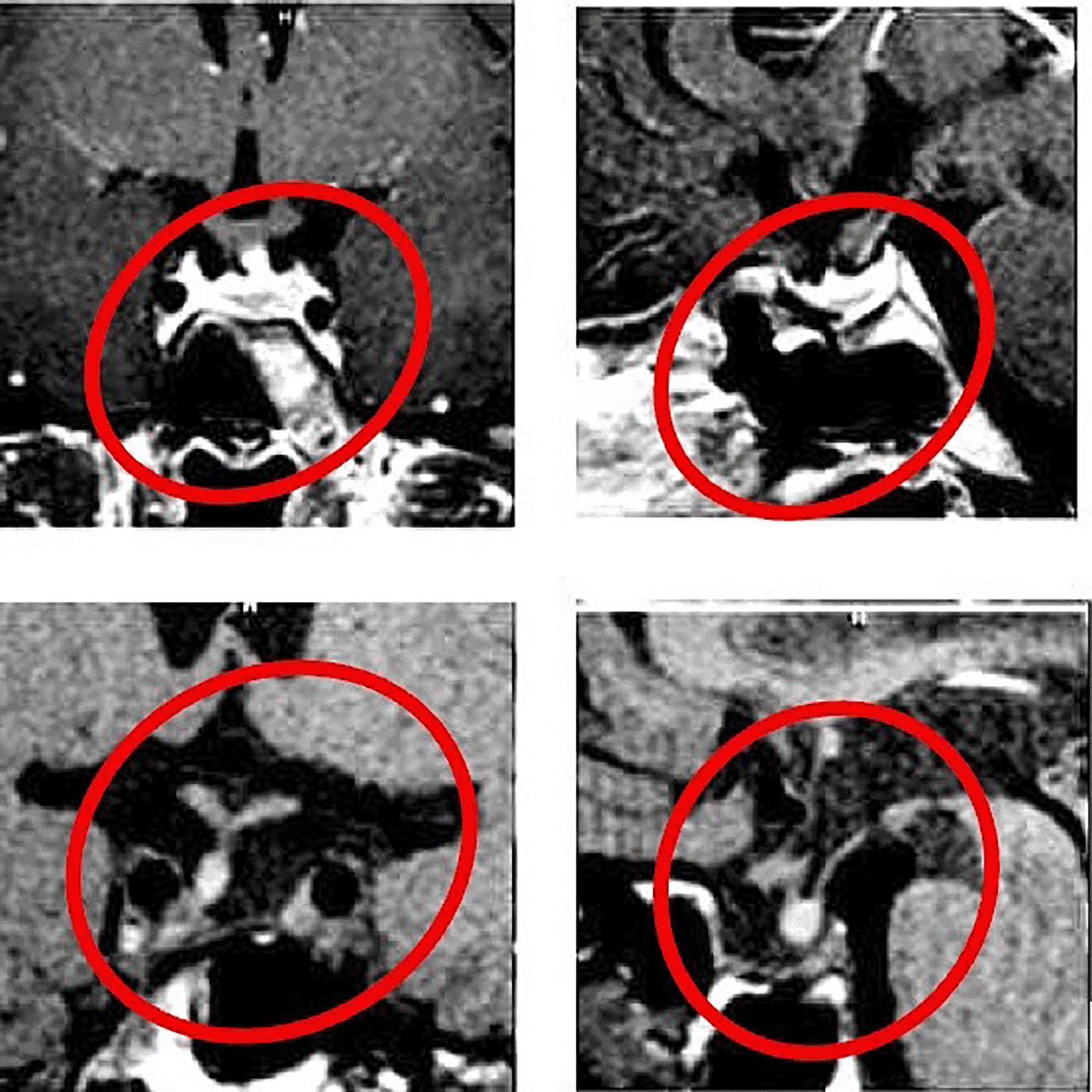
Investigations and Findings: The investigations revealed significant abnormalities, as summarized in Table 1. Key findings included elevated serum calcium and iPTH levels in both cases, imaging-confirmed parathyroid adenomas, and additional biochemical markers supporting the diagnosis of MEN 1. Upper G I Endoscopy showing multiple sessile gastric and duodenal polyps ( Figure 1), Key findings included elevated serum calcium, high iPTH levels, and imaging confirming a left lower parathyroid adenoma ( Figure 2) and MRI Brain showing pituitary adenoma (Figure 3).
| Investigation type | Case 1: A 47-Year-Old Female | Case 2: A 26-Year-Old Female |
|---|---|---|
| Hematological Findings |
|
|
|
| |
| Biochemical Findings |
|
|
|
| |
| Renal and Electrolyte Findings |
|
|
| Imaging Findings |
|
|
|
| |
| ||
| Trends in Biochemical Parameters |
|
|
|
|
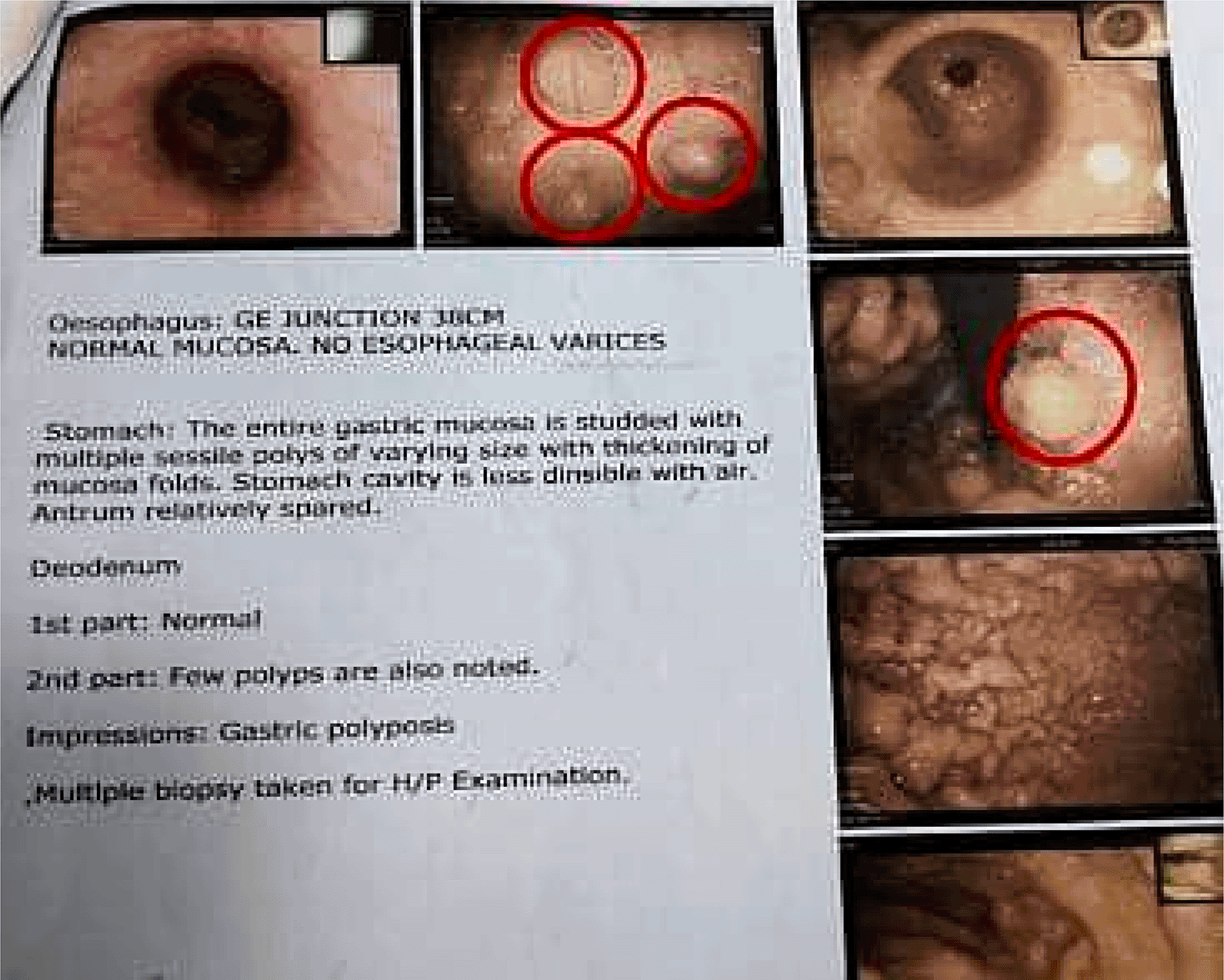
Management: The patient was started on medical therapy due to her poor general condition and advanced CKD.
Parathyroid tablet (30 mg BD): To manage hypercalcemia and suppress parathyroid hormone (PTH) secretion.
Surgery was deferred due to her poor general condition. After a week of medical management, significant improvements were observed, as detailed in Table 1.
Figures
A 26-year-old female presented with a one-month history of persistent headaches and lower limb pain. She had been evaluated at an external center, where imaging revealed multiple osteolytic lesions in the skull. Fine-needle aspiration cytology (FNAC) of one of these lesions demonstrated no evidence of malignancy. She also reported generalized weakness and unintentional weight loss over the past few weeks.
On physical examination, the patient appeared pale with diffuse tenderness over long bones. Neurological assessment was unremarkable, and no focal deficits were noted.
Investigations and Findings
The diagnostic workup revealed significant biochemical abnormalities, strongly indicative of MEN1 syndrome.
• Elevated Serum Calcium: 14 mg/dL (hypercalcemia).
• iPTH Levels: 855 pg/mL (elevated).
• 24-Hour Urinary Calcium Excretion: 398 mg/day (elevated).
• Bone Mineral Density Testing: Severe osteoporosis with a T-score of -4.4.
Imaging Findings:
• MRI Skull ( Figure 4): Demonstrated multiple osteolytic lesions, consistent with skeletal manifestations of MEN1.
• Tc99 MIBI Scan (TECHNETIUM TC 99M SESTAMIBI)TM ( Figure 5): Localized a metabolically active lesion in the right upper parathyroid gland, confirming parathyroid adenoma.
• MRI Abdomen ( Figure 6): Revealed a focal cystic lesion measuring 13x12 mm in the pancreatic tail, consistent with a pancreatic neuroendocrine tumor.
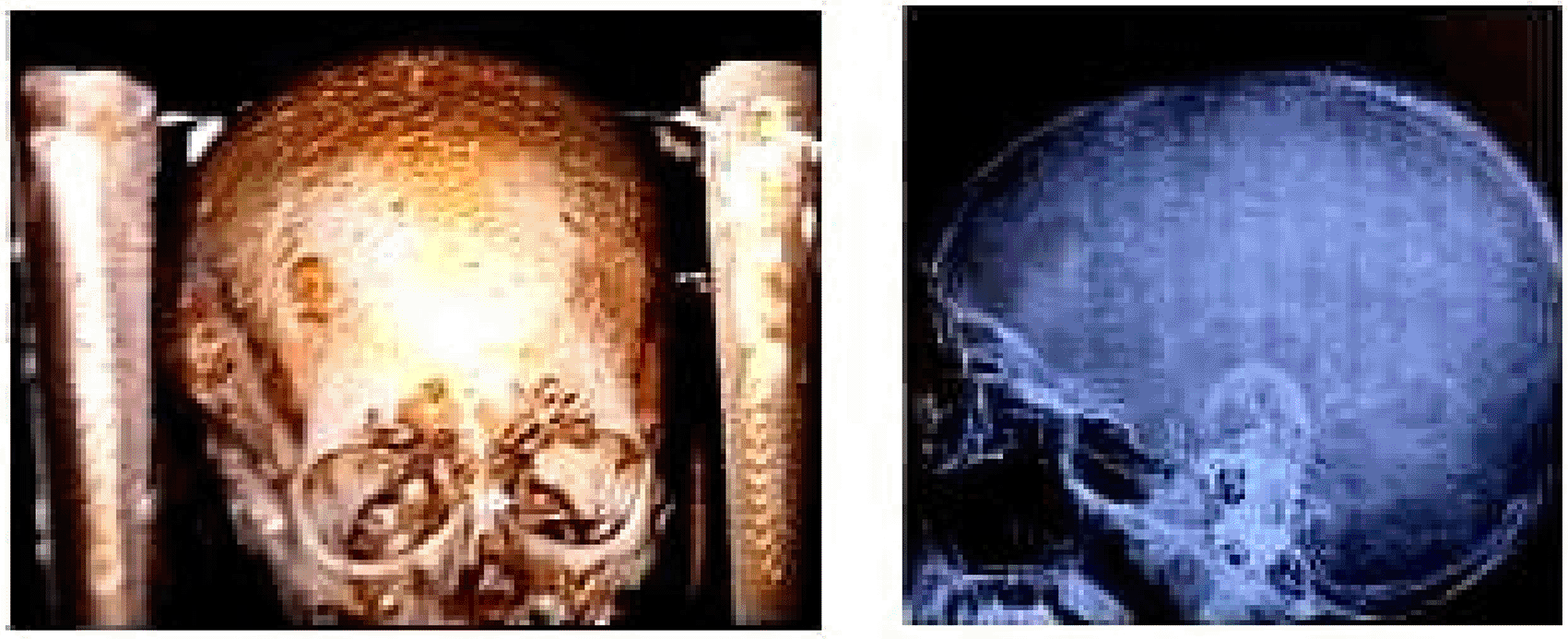
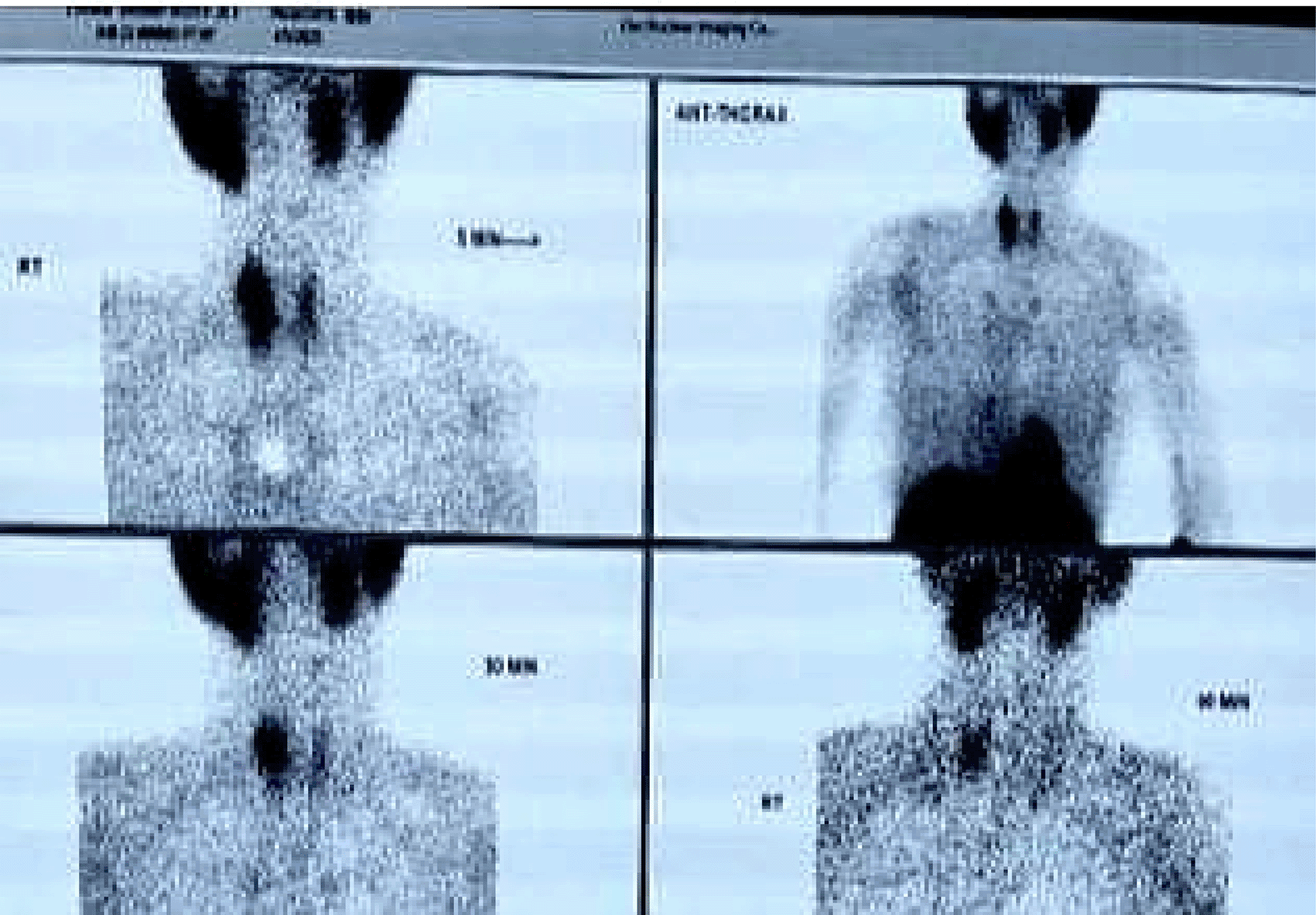
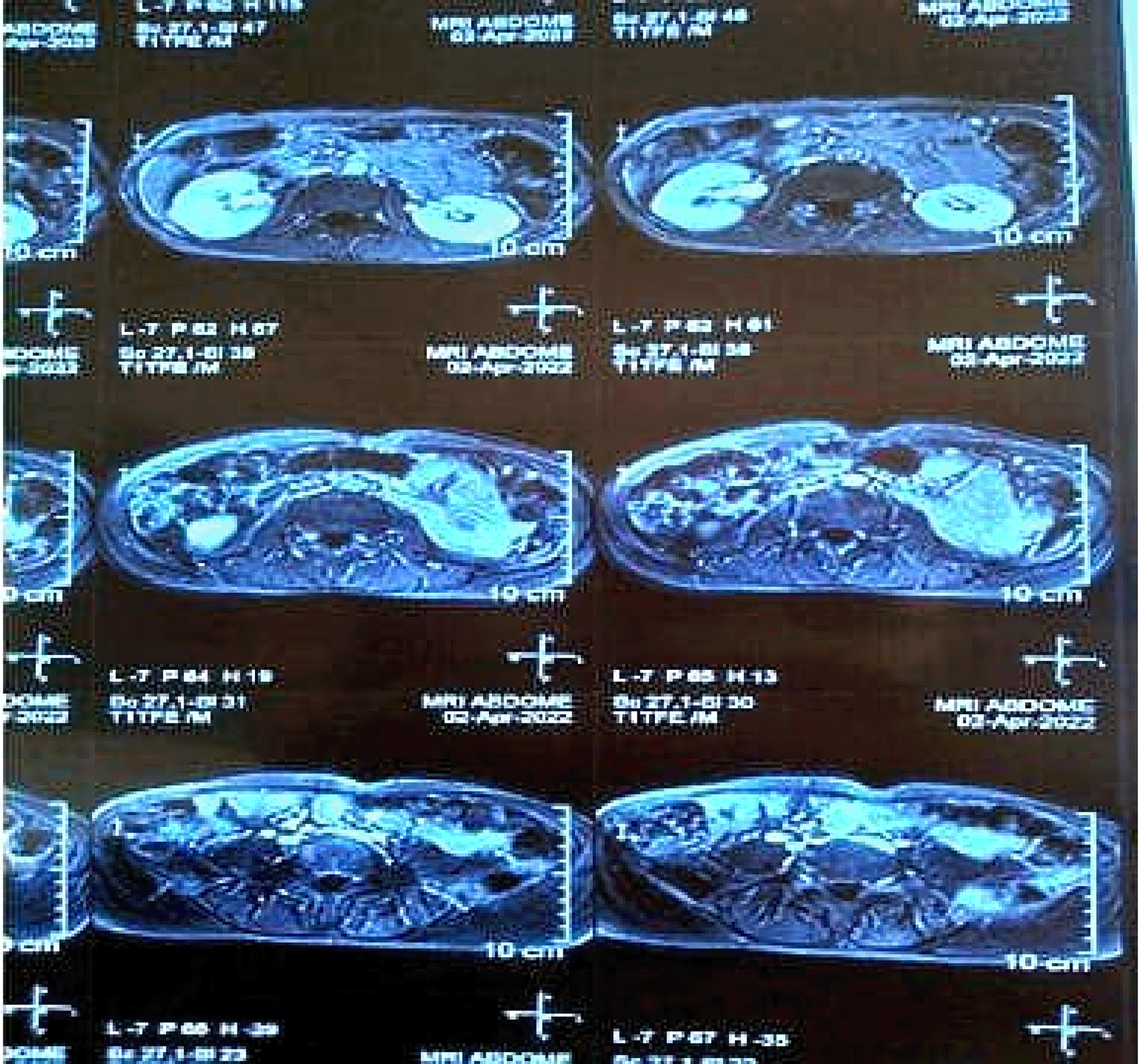
These findings underscored the multisystem involvement characteristic of MEN1, integrating features of hyperparathyroidism and neuroendocrine tumors. A detailed summary of these findings is provided in Table 1.
Management
The patient was initially stabilized for a hypercalcemic crisis with:
1. Intravenous Fluids: To address dehydration and hypercalcemia.
2. Steroids: To assist in calcium homeostasis.
3. Conservative Management of Fractures: Addressed femoral fracture without surgical intervention.
Surgical Intervention
The patient underwent a parathyroidectomy, after which:
• Postoperative Serum Calcium: Dropped to 4 mg/dL and stabilized at ~8 mg/dL with the following:
• Serum iPTH Levels: Decreased dramatically to 5 pg/mL post-surgery, reflecting the successful removal of the hyperactive parathyroid adenoma.
• Thazide diuretics: Although thiazides are generally contraindicated in hypercalcemia, in this patient they were initiated postoperatively to manage persistent hypercalciuria. Their use was carefully guided by close biochemical monitoring and clinical necessity.
Diagnosing MEN1 requires integrating clinical symptoms, biochemical findings, and imaging results to identify the involvement of multiple endocrine organs. In both cases, elevated serum calcium and iPTH levels were pivotal in diagnosing primary hyperparathyroidism, the most common manifestation of MEN1. Imaging modalities like Tc99 MIBI scans and MRI confirmed the presence of parathyroid adenomas and associated neuroendocrine tumors.
The diagnostic process necessitated a multidisciplinary approach:
1. Endocrinology: Managed hyperparathyroidism through biochemical monitoring and medical therapies, including cinacalcet and octreotide.
2. Surgery: Performed parathyroidectomy and provided postoperative care to normalize calcium and PTH levels.
3. Radiology: Utilized advanced imaging techniques to localize adenomas and detect skeletal and abdominal abnormalities.
4. Oncology: Investigated neuroendocrine tumor markers such as chromogranin A and gastrin, guiding further treatment plans.
5. Nephrology: Monitored renal function, particularly in Case 1, where CKD complicated management.
6. Genetic analysis could not be performed due to the unavailability of testing facilities at our center. Both cases had insignificant family history and were thus considered sporadic. Nevertheless, we emphasize the importance of thorough family history assessment, genetic counseling, and genetic testing where feasible, given their implications for early detection and management in MEN1.
The combined expertise of these specialties ensured accurate diagnosis, effective treatment, and comprehensive care, highlighting the importance of collaborative management in MEN1 cases.
Multiple Endocrine Neoplasia Type 1 (MEN1) is a rare autosomal dominant hereditary disorder characterized by tumors in multiple endocrine organs. It arises from mutations in the MEN1 gene, which encodes the tumor suppressor protein menin. Loss of menin function disrupts cellular growth control, resulting in the development of both endocrine and non-endocrine tumors. This discussion addresses the clinical complexities, diagnostic challenges, and therapeutic strategies associated with MEN1, drawing on the insights provided by the two cases presented.5
The MEN1 gene mutation results in the loss of menin function, facilitating unchecked proliferation in endocrine tissues. MEN1 primarily affects the parathyroid glands, pancreas, and anterior pituitary, though other tissues may also be involved. Hyperparathyroidism is the most common manifestation, present in over 90% of cases, and is often the earliest clinical feature. Tumors in the pancreas and gastrointestinal tract, such as gastrinomas and insulinomas, contribute significantly to morbidity. Pituitary adenomas, observed in about 30-40% of cases, frequently cause hormonal imbalances but may also be asymptomatic.5,6
In Case 1, hyperparathyroidism and a gastrointestinal neuroendocrine tumor (gastrinoma) were evident, consistent with MEN1’s hallmark presentations. In Case 2, the combination of skeletal involvement, pancreatic tail lesion, and hyperparathyroidism underscored the syndrome's multisystemic nature.
The diagnosis of MEN1 often requires integrating clinical, biochemical, and imaging findings. Symptoms may vary widely depending on the organs involved, and patients may initially present with vague or nonspecific complaints.
Biochemical Markers
Elevated calcium and intact parathyroid hormone (iPTH) levels are critical in diagnosing primary hyperparathyroidism. Additional markers, such as chromogranin A and gastrin, aid in identifying neuroendocrine tumors. In both cases, these markers provided pivotal diagnostic insights. For instance, the markedly elevated chromogranin A and fasting serum gastrin in Case 1 confirmed the presence of a gastrinoma.
Imaging Modalities
Advanced imaging techniques play a central role in localizing tumors and guiding management. Tc99 MIBI scans are highly effective for identifying parathyroid adenomas, as demonstrated in both cases. MRI and CT imaging further delineate the extent of organ involvement and assess for metastatic disease. In Case 2, MRI revealed osteolytic skull lesions and a pancreatic tail neoplasm, underscoring the systemic involvement of MEN1.7
Genetic Testing
While genetic confirmation of MEN1 is definitive, it is often not immediately necessary for clinical diagnosis. Genetic testing, however, has significant implications for family screening and early detection in asymptomatic carriers. Both cases could benefit from genetic counseling to address familial risk.
The multisystem nature of MEN1 requires a comprehensive and multidisciplinary approach to management, involving endocrinologists, surgeons, radiologists, and oncologists. Each manifestation demands targeted therapeutic strategies:
Hyperparathyroidism
Primary hyperparathyroidism is the most common and earliest manifestation of MEN1. Parathyroidectomy is the treatment of choice, particularly when symptomatic hypercalcemia or significant bone involvement is present. Case 2 exemplifies the success of this approach, with biochemical normalization post-surgery. Medical therapy with cinacalcet can be valuable in patients unfit for surgery, as in Case 1.
Neuroendocrine Tumors
Neuroendocrine tumors (NETs) in MEN1 often secrete bioactive peptides, leading to distinct clinical syndromes. Gastrinomas, as seen in Case 1, can result in severe peptic ulcer disease. Management includes proton pump inhibitors, somatostatin analogs like octreotide, and, where feasible, surgical resection. Pancreatic NETs, such as the lesion identified in Case 2, may require surgery or targeted therapies, depending on the tumor’s functional status and metastatic potential.
Pituitary Involvement
Pituitary adenomas in MEN1 may require medical management with hormone suppression therapies or surgical intervention in cases of significant mass effect or hormonal hypersecretion. Although not prominently featured in these cases, regular screening is essential given the high prevalence of pituitary tumors in MEN1.
Case 1 highlights the challenges of managing MEN1 in the presence of chronic comorbidities like CKD, which complicate surgical and medical interventions. Conservative management with cinacalcet and octreotide successfully stabilized the patient’s biochemical parameters, delaying the need for surgery. This underscores the importance of individualized treatment strategies.
In contrast, Case 2 demonstrates the more aggressive presentation of MEN1 in a younger patient. The presence of severe osteoporosis, hypercalcemic crisis, and pancreatic involvement necessitated prompt surgical intervention. The significant improvement in biochemical parameters post-parathyroidectomy illustrates the efficacy of timely surgical management.
MEN1 is a lifelong condition requiring regular surveillance to detect new or recurrent tumors. Annual monitoring of serum calcium, PTH, chromogranin A, and imaging studies is recommended. Early detection of asymptomatic tumors can significantly improve outcomes. Genetic counseling and testing are crucial for first-degree relatives, as early identification in asymptomatic carriers enables proactive monitoring and intervention.8
Multiorgan Involvement: MEN1 patients frequently develop tumors in multiple endocrine glands, primarily affecting the parathyroid, pancreas, and pituitary glands. Some cases also present with skeletal involvement (osteolytic lesions) or mediastinal masses, underscoring the need for a comprehensive diagnostic approach shown in Table 2.
Diagnostic Markers and Biochemical Evaluation: Elevated serum calcium and intact parathyroid hormone (iPTH) levels are consistent indicators of primary hyperparathyroidism, the most common MEN1 manifestation. For neuroendocrine tumors (NETs), Chromogranin A and Gastrin serve as valuable biomarkers, while fasting insulin and C-peptide levels are crucial for diagnosing insulinomas.
Role of Imaging in MEN1 Diagnosis: A combination of imaging techniques is essential for tumor localization and staging. Tc99 MIBI scans are effective for identifying parathyroid adenomas, whereas MRI and CT scans are preferred for detecting pancreatic and pituitary tumors. PET scans are utilized selectively, particularly in cases of suspected metastatic disease.
Genetic Testing and Family Screening: Given the hereditary nature of MEN1, genetic testing is crucial for confirming diagnoses and screening at-risk family members. Identifying asymptomatic carriers facilitates early detection and preventive care.
Treatment Strategies: Surgery vs. Medical Management: Surgical interventions, such as parathyroidectomy and tumor resection, are standard treatments for MEN1-related tumors. In cases where surgery is not feasible, medical therapies like Cinacalcet and Somatostatin analogs are effective for hormonal control and symptom management.
Follow-up and Long-Term Surveillance: Due to the lifelong risk of tumor recurrence, structured long-term follow-up is essential. Annual biochemical screenings and routine imaging are recommended to detect early tumor recurrence or new growths. Additionally, genetic counseling is vital for at-risk family members to ensure proactive disease management.
The two cases illustrate the diverse clinical spectrum of MEN1, ranging from mild to severe systemic involvement. The complexities of diagnosis and management necessitate a multidisciplinary approach tailored to individual patient needs. Advances in biochemical and imaging diagnostics, coupled with targeted medical and surgical therapies, continue to enhance outcomes for patients with this challenging syndrome. Long-term surveillance and genetic counseling remain pivotal in managing MEN1 and preventing complications in affected families.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)