Keywords
Myelocystocele, Spina bifida, Saccrocoygeal teratoma, cerebrospinal fluid
Myelocystocele, Spina bifida, Saccrocoygeal teratoma, cerebrospinal fluid
Congenital anomalies are significant contributors to both morbidity and mortality in children. The effects of these anomalies extend beyond the individual, placing a burden on healthcare systems and families. Congenital central nervous system (CNS) anomalies, specifically neural tube defects (NTDs), are classified as either open or closed NTDs based on the presence or absence of exposed neural tissue.1 Skin-covered terminal myelocystoceles (TMC) represent a clinical entity characterized by turbulent cerebrospinal fluid (CSF) drainage, an associated caudally elongated spinal cord, terminal cystic dilation of the dorsal spine, and an intact dura mater.2,3 Several malformations have been associated with TMC, affecting the anorectal region, genitourinary system, and vertebrae.4
Skin-covered malformations encompass a wide range of spinal dysraphism entities, such as spinal cord meningoceles or lipomas, making clinical and radiological differentiation between these conditions crucial for choosing appropriate management approach and avoiding associated neurological sequelae, such as incontinence and motor weakness.4,5 In this case report, we describe our experience managing a neonate presenting with TMC. An informed consent was taken from the patient guardian. To the best of our knowledge, there are no previous reported TMC cases in Saudi Arabia.
A two-week-old male neonate, born to a 19-year-old primigravida, para 1 mother, was referred to the neurosurgery department for evaluation of a giant swelling in the lower back. The neonate was born full term via spontaneous vaginal delivery, which was uneventful. There was no significant medical or surgical history, except for the mother’s hypothyroidism, for which she was not on regular treatment. No infections were noted during the gestational period, and neither parent has a history of diabetes mellitus.
A prenatal anatomical ultrasound screening at 20 weeks revealed sacrococcygeal anomalies. At birth, the neonate cried spontaneously, with an APGAR score of 8 at both 1 and 5 minutes. The neonate weighed 2745 grams, measured 54 cm in length, and had a head circumference of 33 cm. There was no noted CSF leak.
On examination, the neonate was active and alert. A subcutaneous light bluish cystic swelling was noted in the midline of the lower lumbosacral region ( Figure 1). The swelling size was observed to increase during crying, and the transillumination test was positive. The head circumference and spine curvature were normal; the plantar reflex was positive, and tone changes were unremarkable. The anterior fontanelle was open and flat, and no other associated abnormalities were noted.
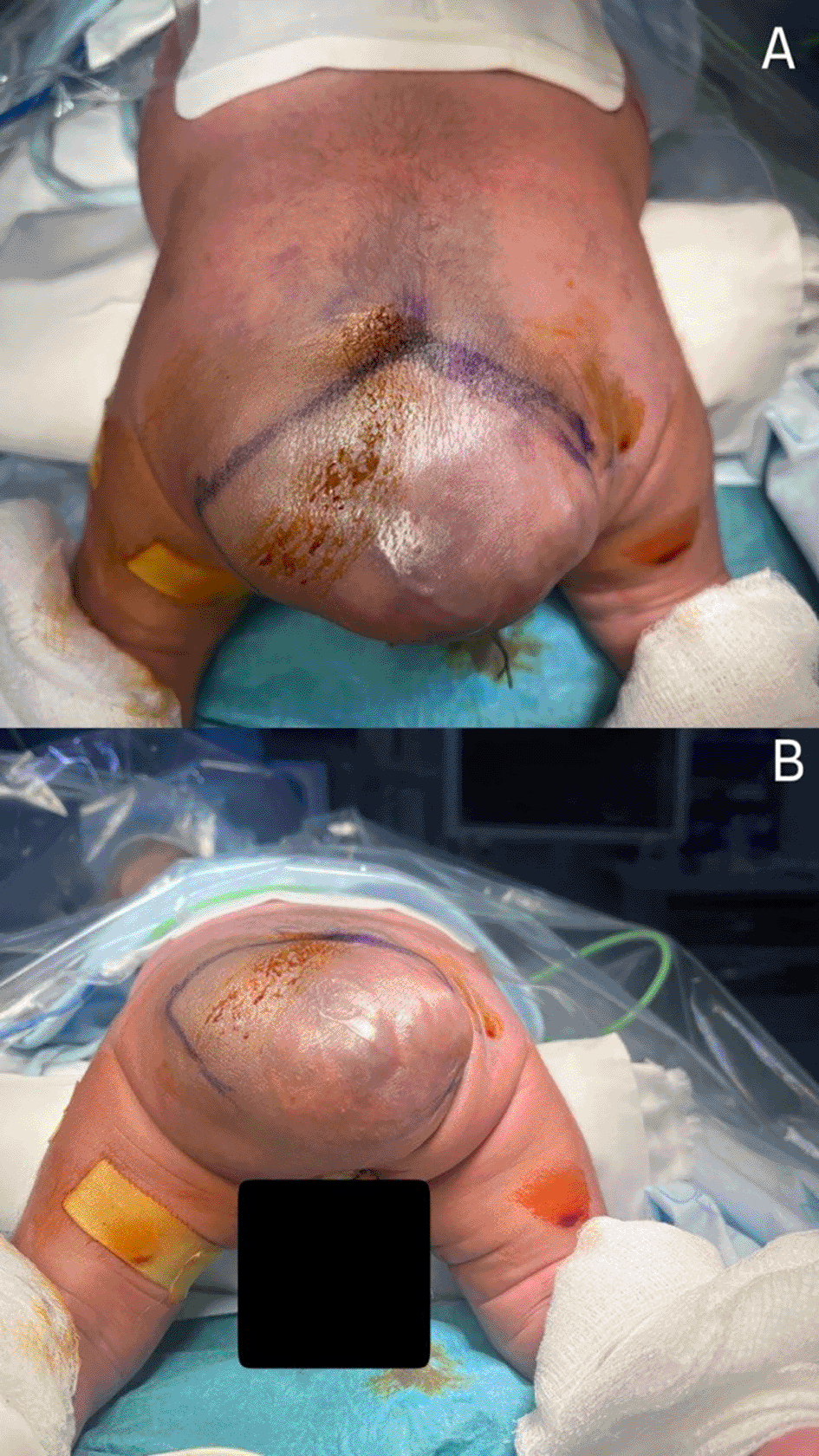
The edema results in considerable occlusion of the intergluteal cleft and presents as smooth and fluctuant upon examination and palpation. The lesion exhibits no discernible hard material, and its bluish coloration indicates a delicate covering skin potentially concealing a fluid accumulation beneath. The adjacent skin is intact, exhibiting no symptoms of erythema or inflammation, and there is an absence of discharge or ulcers.
Several vertebrae demonstrated spina bifida on plain radiographs. MRI of the spine revealed a well-defined sacrococcygeal exophytic multilobulated cystic lesion ( Figure 2). Based on these findings, our preliminary diagnosis was either a pure cystic sacrococcygeal teratoma or a TMC.
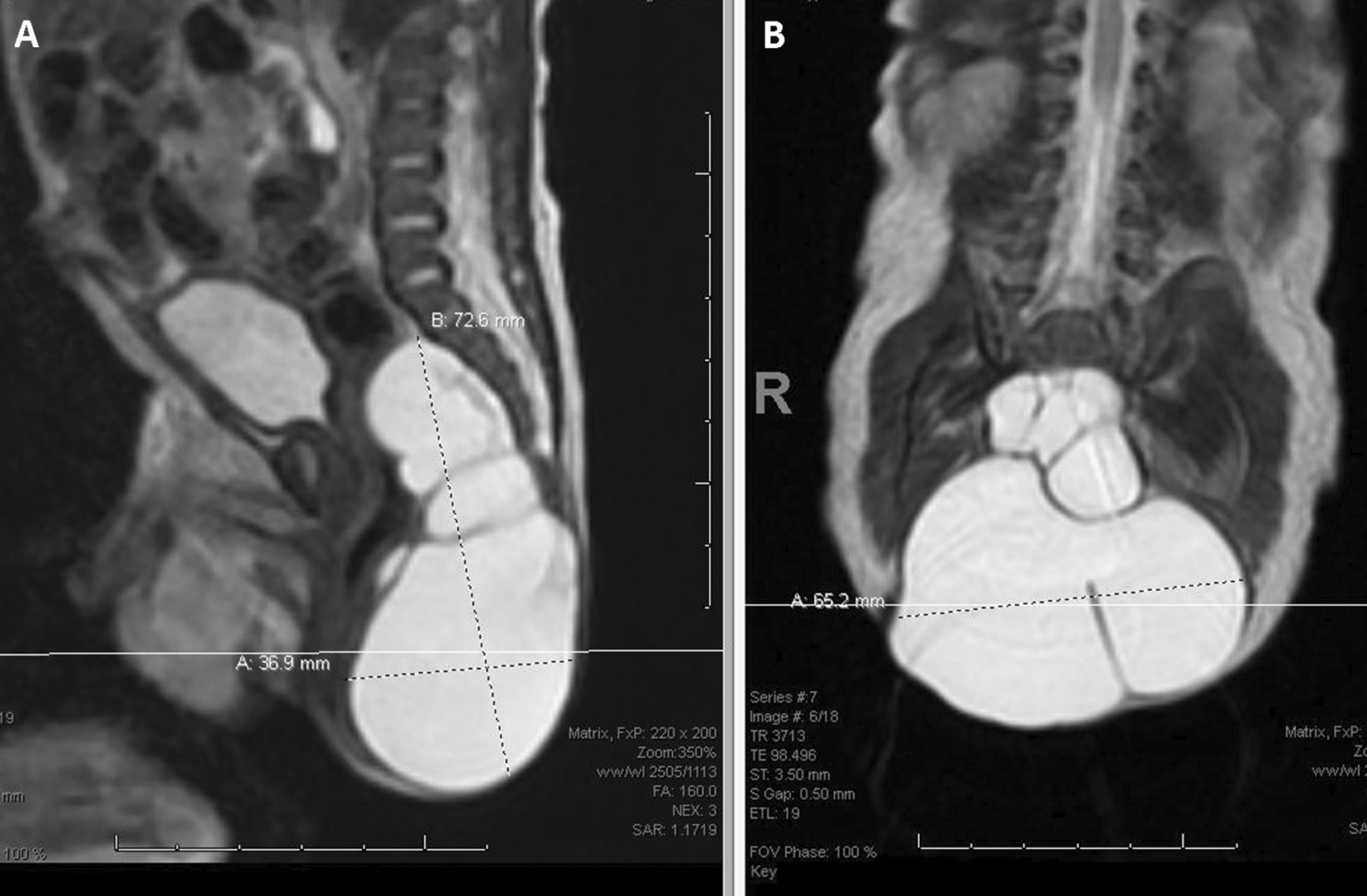
The lesion arises from an anterior sacral defect, exhibiting herniation of the meningeal sac through the sacral foramen. It spreads into the pelvic cavity and measures approximately 3.4 × 6.6 x 7.2 cm (anterior-posterior x transverse x craniocaudal dimension). A bulk effect is observed on the rectum and left ureter. No indication of adipose tissue, calcification, or soft tissue component is observed.
Surgical excision was performed through two inverted V-shaped incisions along the caudal region of the mass. The dura mater was exposed along the edges of the swelling through lateral skin dissection. The meningocele sac was found to continue terminally with the spinal subarachnoid space, and the inner thin-walled sac was found to terminate in the spinal canal. Both were opened and drained. The dura mater was primarily reconstructed, followed by multilayered skin closure ( Figure 3).
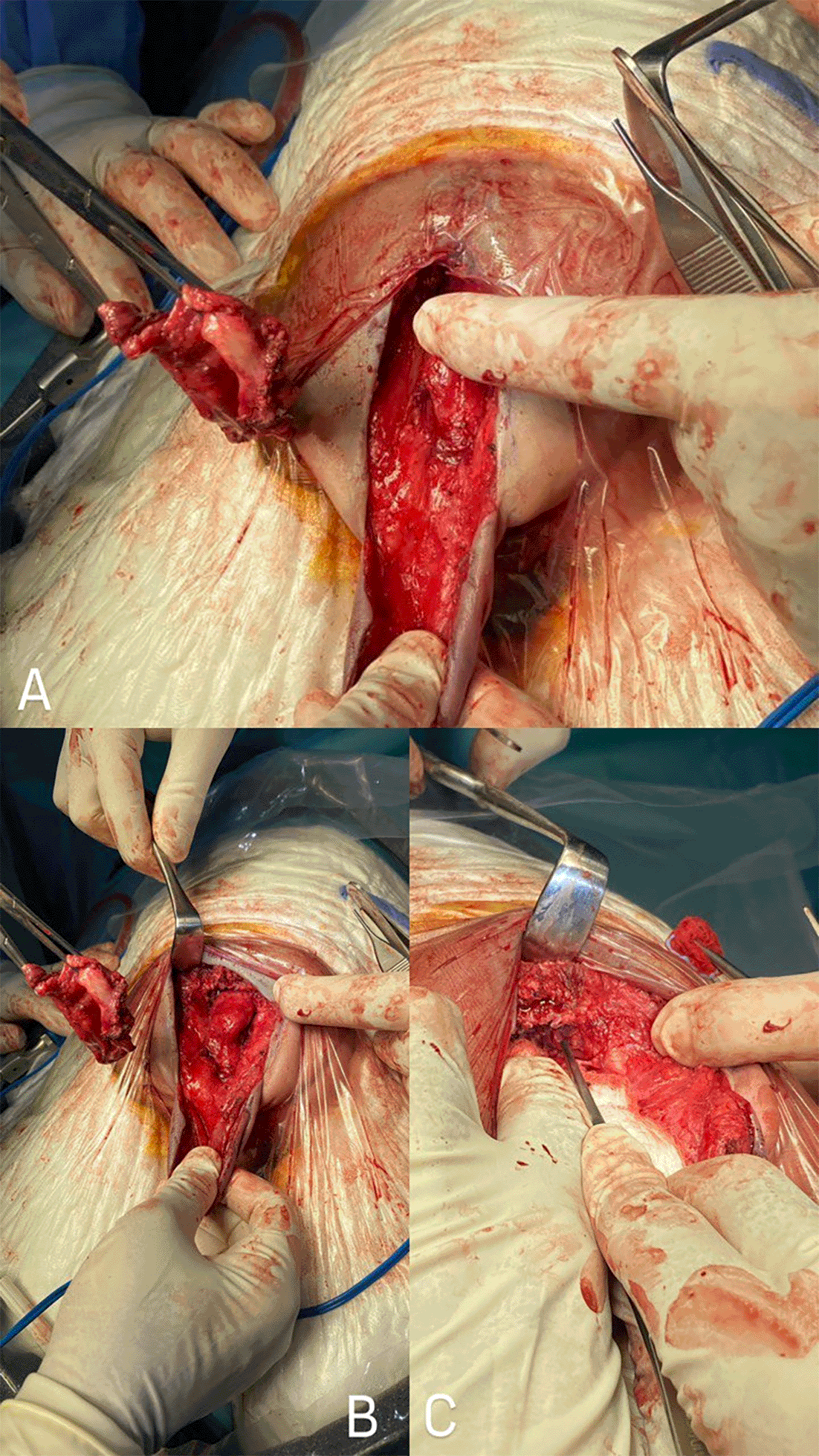
A. Exposing the inner lining of the sac following the incision of the skin and cystic wall, with subsequent fluid drainage. B. Detaching and removing the sac, including its adherent attachment to the coccyx. C. Removing the coccyx along with the sac, resulting in exposure of the posterior rectal wall.
Postoperatively, the patient was nursed prone for a week. Intravenous antibiotics were administered for one week. Sutures were removed two weeks after discharge. After 8 months of outpatient follow-up, the baby remained free of neurological impairments.
Myelocystocele is an uncommon closed NTD characterised by the herniation of the enlarged spinal cord central canal through a posterior spinal defect.6,7 In contrast to teratomas, which comprise tumours of all germ layers, myelocystoceles are non-neoplastic entities. They contrast with myelomeningocele, in which the spinal cord and meninges protrude without the development of a genuine central canal dilatation.6
Myelocystoceles has an incidence of approximately 4 to 5 per 10,000 live births, accounting for a portion of the estimated 140,000 NTDs that occur globally each year with.8,9 These may present as bowel and bladder problems, tethered cord syndrome, and orthopaedic anomalies such as, scoliosis. The condition is frequently associated with additional congenital anomalies such as, the OEIS complex (omphalocele, bladder exstrophy, imperforate anus, and sacral agenesis).
Neurological manifestations are contingent upon the degree of spinal involvement.9 Myelocystocele entails the atypical closure of the neural tube during embryonic development, generally occurring between days 22 and 28 of gestation. Deficiencies in the folding or fusing processes result in anomalies in the spine or skull. Environmental variables, such as maternal folate insufficiency, and genetic abnormalities are associated with the disruption of neural tube development.10 An antenatal ultrasound usually shows a cystic tumour across the lower spine. A “cyst within a cyst” look, with a thin-walled cyst inside a bigger cyst, is characteristic. Anechoic masses with thin walls rarely have solid tissue, calcification, or intrapelvic extension. The foetal head often reveals a normal posterior fossa without Chiari II malformation (e.g., no ventriculomegaly or “banana sign”). The OEIS complex can cause myelocystoceles and spinal dysraphism.7
Diagnosis is typically made shortly after birth by physical examination. While some neonates present with normal neurological findings, many exhibits clinical signs such as, cystic mass during physical examination or upon detailed urological or electrophysiological assessment at the time of diagnosis. Consequently, surgical intervention such as, excision of the cyst, repair of the spinal defect, de-tethering of the spinal cord is recommended even for asymptomatic individuals.11
In our instance, plain radiographs revealed spina bifida in several vertebrae. An identifiable sacrococcygeal exophytic multilobulated cystic lesion with internal septations was observed on the spine's MRI. Our first diagnosis was either a TMC or a purely cystic sacrococcygeal teratoma.
Two inverted V-shaped incisions were performed in the caudal region of the mass to facilitate surgical removal. A key clinical consideration in the management of TMC is the potential for cyst expansion, which serves as an indicator of rapid neurological decline. For example, the development of foot deformities within a two-month period reflects how quickly deterioration can occur. This progression is believed to result from the cyst’s expansion, which may cause direct stretching of the spinal cord and adjacent neural tissues dorsally. Therefore, caregivers should be informed about the risk of cyst enlargement and advised to seek immediate medical attention if such growth is observed.12
Myelocystocele is an uncommon yet critical neural tube condition that requires prompt identification, comprehensive diagnostic assessment, and appropriate treatment to avert enduring neurological and functional consequences. Surgical intervention, encompassing cyst excision, spinal defect correction, and spinal cord de-tethering, is fundamental to reduce the danger of fast neurological deterioration. Holistic treatment approaches, encompassing rehabilitation and familial education, are essential for enhancing functional results and quality of life for impacted persons.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)